Mitochondria and the endoplasmic reticulum (ER) physically and functionally interact, regulating each other’s function, but the molecular mechanisms remain not fully understood. In this study, we revealed that ER-associated degradation (ERAD), an ER protein quality control mechanism, governs ER Ca2+ entry into mitochondria by mitochondria-associated ER membrane (MAM) in hepatic cells. Inhibition of ERAD by pharmaceuticals or genetic ablation of the key ERAD protein SEL1L resulted in altered mitochondrial morphology, reduced mitochondrial energy production, and increased Ca2+ transfer from ER to mitochondria. Additionally, SEL1L absence caused an increase in the number of MAM. In ERAD-deficient hepatic cells, a reduction in the number of MAM or knockdown of the inositol 1,4,5-trisphosphate receptor (IP3R), which is responsible for ER Ca2+ release, partially restored mitochondrial Ca2+ signaling and bioenergetics. Together, these results suggest that ERAD plays a crucial role in regulating mitochondrial bioenergetics, suggesting the potential to improve cellular and organismal mitochondrial function by increasing cellular ERAD activity.
Research Article
ERAD deficiency disrupts mitochondrial bioenergetics by altering MAM Ca2+ in human hepatic cells
https://doi.org/10.21203/rs.3.rs-4939621/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version

hepatic cells
ERAD
mitochondrial function
MAM
mitochondrial calcium
Interorganelle communication occurs through membrane contact sites and is crucial for the maintenance of organelle homeostasis and organismal health (Arora et al. 2022; Henne et al. 2021;Wu et al. 2018). Interactions between endoplasmic reticulum (ER) and mitochondria are excellent examples of the importance of this partnership. The close physical contact (~ 30 nm) between the ER and mitochondria occurs in a region known as the mitochondria- associated ER membrane (MAM). MAM effectively integrates signal transduction with metabolic pathways to regulate ER-mitochondrial communication. It plays important regulatory roles in several key cellular processes including Ca2+ exchange, lipid synthesis, ER stress (ERS), mitochondrial dynamics and function, autophagy, and apoptosis (Rowland et al. 2012; Phillips et al. 2016; Jang et al. 2022). Evidence indicates that dysfunction in MAM contributes to conditions such as neurodegenerative diseases, fatty liver disease, and diabetes (Liu et al. 2022; Beaulant et al. 2022; Elwakiel et al. 2024). There is still considerable uncertainty regarding how interorganelle communication operates and the physiological consequences of miscommunication.
Quality control systems normally maintain homeostasis and proteostasis in the ER throughout an organism's lifespan, and ER-associated degradation (ERAD) is an important branch. The primary function of ERAD is to identify, sort, and degrade misfolded proteins to prevent the harmful effects caused by their buildup. This is particularly important in secretory cells (Kang et al. 2021). The Sel1L-Hrd1 protein complex represents the most highly conserved branch of ERAD from yeast to humans. Recent studies have found that the Sel1L-Hrd1 ERAD may have a constitutive function in maintaining optimal concentrations of key proteins that control mitochondrial dynamics (Zhou et al. 2020). However, little is known about the pathophysiological effects of defective ER quality control systems on mitochondrial function and vice versa in secretory cells such as hepatocytes.
We had previously shown that disrupting ERAD genetically and pharmacologi- cally impairs glucose-stimulated insulin secretion in β-cells in vitro and in mice (Hu et al. 2019). In the current study, we investigated the impact of ERAD deficiency on mitochondria both structurally and energetically using an ERAD-deficient hepatic cell line model. The consequences of ERAD deficiency were linked to aberrant MAM levels, which, in turn, caused mitochondrial Ca2+ excess. Our results indicate that ERAD is critical for maintaining normal mitochondrial energy production and raise the potential for enhancing cellular ERAD capacity to improve mitochondrial function at the organismal and cellular levels.
2.1. Cell culture and treatment
HepG2 cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) supplemented with 10% FBS(Gibco) and 1% penicillin/streptomycin in a 5% CO2 incubator at 37°C. The cells were treated with ERAD inhibitor eeyarestatin I (8 µM; MedChemExpress, USA), cycloheximide (50 µg/mL; Abmole, China), 2-APB (20 µM; Sigma-Aldrich, USA), ATP (10 µM; MedChemExpress), palmitic acid (0.5 mM; Sigma-Aldrich), si-con or siIP3R (10 nM; Gemma-Pharma, China) for the corresponding times. Cells between passages five and eight were used for all experiments.
2.2. CRISPR/Cas9 mediated Sel1l gene editing in HepG2 cells
Cas9-mediated editing of Sel1l was performed as previously described (Liu et al. 2020). Briefly, sgRNAs flanking exon 6 were designed using the Optimized CRISPR Design tool (http://crispr.mit.edu/) and cloned into the expression vector pGL3- U6 -2sgRNA. The sgRNA expression vector and Cas9 expression vector pST1374-NLS-3xFlag- linker-Cas9 were both inserted into HepG2 cells using Lipofectamine 3000 (Baillat et al. 2016). After transfection, 5 µg/mL of puromycin was added to the growth medium to specifically choose cells that are not killed by puromycin. Approximately one week later, puromycin-resistant HepG2 colonies were selected, expanded, and tested by PCR.
2.3. Mitochondrial morphology, mitochondrial membrane potential and ATP production assay
To investigate the effect of ERAD on mitochondria, we examined the mitochondrial morphology, mitochondrial membrane potential, and cellular ATP levels.Mitochondria were visualized using a MitoTracker Green fluorescent probe (Invitrogen, USA) or Tom20, followed by observation with laser confocal microscopy and analysis using ImageJ software.
The mitochondrial membrane potential was determined using a JC-1 assay kit (Beyotime,China). Briefly, HepG2 cells were cultured in 6-well plates at a density of 2x10*5 cells per well and treated as required (EerI, 2APB, or siIP3R). The cells were then incubated with 2 µM JC-1 for 30 min at 37°C in the dark, washed with PBS, and the fluorescence was measured using flow cytometry (BD,USA) and/or a fluorescence microscope (Olympus, JAPAN).
Cellular ATP levels were measured using a Firefly Luciferase ATP Assay Kit (Beyotime) according to the manufacturer’s instructions. Briefly, treated cells were lysed and centrifuged for 5 min at 12000 g to obtain the cell supernatant. After mixing 100 µL of cell supernatant with 100 µL of ATP assay working solution in a 96-well plate, the reaction was allowed to proceed for 5 min at room temperature. The fluorescence intensity was quantified using a microplate reader (Molecular Devices), and the ATP value was normalized to the protein concentration.
2.4. Western blot analysis
As previously mentioned (Sha et al. 2009), cell lysates were prepared and western blotting was performed. Briefly, cells were scraped and resuspended in 10 mM Tris–HCl pH 7.5, 1 mM PMSF, and incubated at 4 ℃ for 20 min. The lysate was centrifuged at 10,000 g for 10 min at 4°C to obtain the supernatant. The protein concentration in the supernatant was determined using a BCA kit (Thermo Fisher, USA). Denaturing buffer was added to the protein supernatant, which was boiled for 5 min before separation by SDS-PAGE. The following antibodies were used in this study: GFP (1:500 rabbit) from Santa Cruz Biotechnology, β-actin (1:1,000 mouse) from Sigma-Aldrich, Sel1L (1:1,000 rabbit) and IP3R (1:500 rabbit) from Abcam. Anti-rabbit or anti-mouse secondary antibodies were incubated for 2 h at room temperature and band density was quantified using ImageJ software.
2.5. Cytosolic, ER and mitochondrial Ca2+ measurement
HepG2 cells were seeded into 6-well glass-bottomed chambers at a density of 1×105 cells/well and cultured in DMEM for 24 h. Ca2+ in the cytoplasm, ER, and mitochondria was detected using Fluo4, AM, Mag-Fluo4, and Rhod2 probes. The probes were then added to Ca2+-free PBS and incubated for 30–60 min at room temperature. Using a fluorescence microscope, fluorescent images were collected every 50 s, background-corrected, and analyzed using the ImageJ software.
2.6. Cell immunofluorescence
HepG2 cells were cultured overnight in a 6 cm dish. After fixation with 4% paraformaldehyde for 15 min, the cells were permeabilized for 5 min with 1% Triton X-100 in PBS. To prevent non-specific binding, cells were incubated in 10% donkey serum in TBST for 1 h at 37°C. Then, primary antibodies calnexin (1:200, Santa Cruz Biotechnology) and Tom20 (1:200, Absin) were introduced to the cells for 1 h at 37 ℃. The cells were then incubated with secondary antibodies in 5% donkey serum for 2 h at room temperature and washed. Stained cells were imaged using laser confocal microscopy, and the intensity and colocalization of fluorescence were analyzed using ImageJ software.
2.7. SiRNA-mediated knockdown of IP3R in HepG2 cells
A scrambled negative control siRNA for hIP3R was predesigned and purchased from Gemma Pharma (China). HepG2 cells were seeded in 6-well plates. When the cells were 80% confluent, Opti-MEM was mixed with either control or IP3R siRNA together with Lipofectamine 3000 (Invitrogen) and incubated for 6 h. After transfection, the medium was replaced with DMEM containing 10% FBS and the cells were cultured for an additional 48 h. Cell lysates were collected to detect IP3R expression levels by western blotting.
2.8. Mitochondrial respiration assay
Mitochondrial respiration was assessed by measuring the oxygen consumption rate (OCR) using a Seahorse XFe96 Analyzer as described previously (Mthembu et al. 2024). HepG2 cells were grown overnight in Seahorse XFe96 microtiter wells at a density of 3×104 cells/well, and treated as required. On the first day of the experiment, the culture medium was replaced with Seahorse XF Base Medium (Agilent Technologies) supplemented with 10 mM glucose, 2 mM glutamine, and 1 mM sodium pyruvate. The following mitochondrial stress test compounds (1 µM Oligomycin, 0.5 µM FCCP, 0.5 µM Rotenone, and 0.5 µM antimycin A) were sequentially injected into the wells at specified time points. The OCR values were normalized to the total protein content in each well and determined using the BCA Protein Assay Kit (Beyotime).
2.9. Statistical analysis
Data are shown as mean ± S.E. Differences between compared groups were evaluated using SPSS software (version 16.0) to perform Student’s t-test or analysis of variance (ANOVA). The significance level was set at P < 0.05.
3.1. ERAD dysfunction impairs HepG2 cells mitochondrial bioenergetics
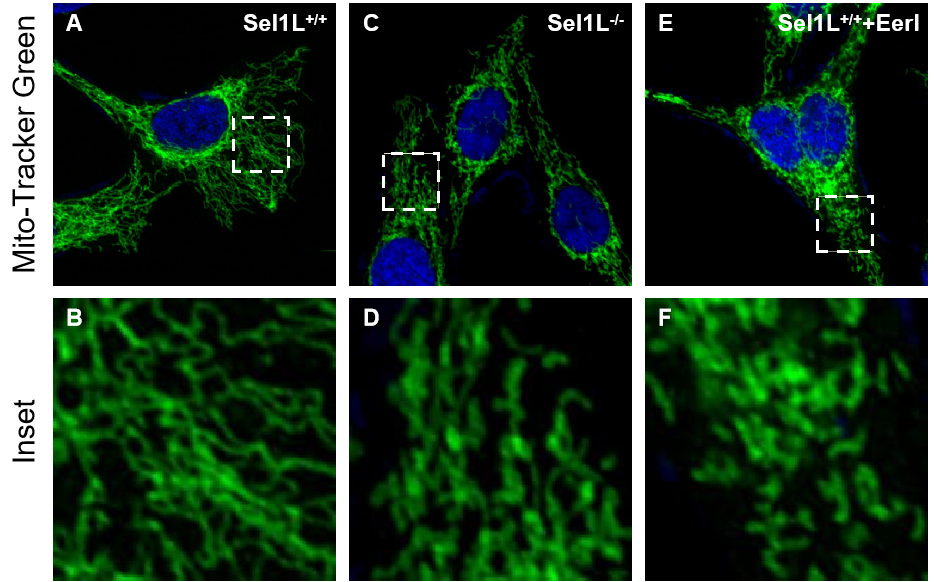
Our previous study showed that disrupting ERAD decreases ATP production in the INS-1 rat pancreatic β-cell line, suggesting that a deficiency in ERAD compromises the mitochondrial respiratory function(Hu et al. 2019). To investigate whether this phenomenon extends to other mammalian cells, we treated human hepatic HepG2 cells with the ERAD inhibitor eeyarestatin I (EerI). EerI treatment reduced ATP levels in a time-and dose-dependent manner (Fig. 1A, B), altered mitochondrial morphology (Fig. 1C-1F, and S1A-F), and mitochondrial membrane potential (MMP) was reduced (Fig. 1G-1L).
SEL1L is essential for the formation of the ERAD complex on the ER membrane in mammals. To further investigate the effect of ERAD on ATP levels, we used CRISPR /Cas9-based gene editing to generate SEL1L-deficient (Sel1L−/−) HepG2 cells (Fig. 2A, B). Next, we used cycloheximide (CHX)-based pulse-chase analysis for α1-antitrypsin null Hong Kong variant (NHK) in WT and Sel1L−/− HepG2 cells to determine the ERAD-degrading function. As shown in Fig. 2C and 2D, the folding-deficient mutant NHK displayed efficient degradation in WT HepG2 cells, but not in Sel1L−/− HepG2 cells. This result indicates that the ERAD deficiency cell model was successfully constructed using the sel1L knockout. For ATP and MMP, Sel1L−/− HepG2 cells were lower than WT HepG2 cells (Fig. 2E, F). Overall, our findings suggest that ERAD deficiency (treatment with EerI or deletion of Sel1L) impairs mitochondrial morphology and bioenergetics in hepatic cells.
3.2. ERAD deficiency causes increased mitochondrial Ca2+ mainly originated from ER
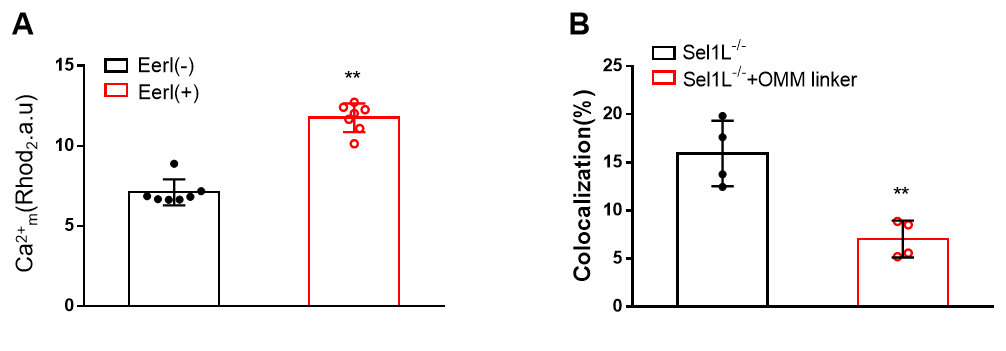
It is well established that mitochondrial Ca2+ concentration is a critical determinant in mediating mitochondrial respiration (Sun et al. 2023; Santulli et al. 2015). Given our findings in pancreatic β-cells that ERAD deficiency causes changes in mitochondrial Ca2+ levels (Hu et al. 2019), we tested whether a similar change occurs in ERAD-defective HepG2 cells. In ERAD-defective HepG2 cells, we used the fluorescent probe Rhod2 AM to detect mitochondrial Ca2+. As shown in Fig. 3A, B, and S2A, both Sel1L deletion and EerI treatment produced a significant increase in Ca2+ levels in the mitochondria of HepG2 cells.
Since ERAD is an ER-associated degradation mechanism, and the ER is a major Ca2+ storage reservoir in eukaryotic cells (Christianson et al. 2023), we investigated whether ER Ca2+ contributes to increased mitochondrial Ca2+ in ERAD-deficient cells. First, we measured ER Ca2+ using the Mag-Fluo4 probe and found that ER Ca2+ levels in Sel1L−/− cells were higher than in WT cells (Fig. 3E, F). Next, we treated WT and ERAD-deficient cells with the inositol 1,4,5-trisphosphate receptor (IP3R) inhibitor, 2APB, which blocks ER Ca2+ release. As shown in Fig. 3C and 3D, ERAD-deficient cells treated with 2APB showed a marked reduction in mitochondrial Ca2+ levels. When stimulated with ATP, which promotes the release of ER Ca2+, ERAD-deficient cells displayed higher mitochondrial Ca2+ uptake than the WT cells (Fig. 3G). Upon addition of 2APB, there was a significant and abrupt decrease in mitochondrial Ca2+ levels in both ERAD-deficient and WT cells (Fig. 3G). Together, these findings indicate that increased mitochondrial Ca2+ originates mostly from the ER in ERAD-deficient hepatic cells.
3.3. ER Ca2+ enters mitochondria via the MAM connections in ERAD-deficient cells.
To evaluate whether increased mitochondrial Ca2+ is the result of direct transport through MAM junctions or IP3R-mediated ER Ca2+ release into the cytosol, followed by mitochondrial uptake, we analyzed cytosolic Ca2+ dynamics in response to IP3R stimulation in WT and Sel1L−/− HepG2 cells using Fluo4-AM. The baseline cytosolic Ca2+ concentration was significantly (P < 0.05) higher in Sel1L−/− HepG2 cells (Fig. 4A). However, ATP stimulation caused a comparable increase in cytosolic Ca2+ levels in WT and Sel1L−/− HepG2 cells (Fig. 4B), despite the higher mitochondrial Ca2+ peak in ERAD-deficient cells (Fig. 3G, 4C). These findings suggest that ERAD deficiency leads to increased Ca2+ transport from the ER through MAM connections, resulting in elevated mitochondrial Ca2+ in HepG2 cells.
To investigate how MAM affects Ca2+ fluctuations between the ER and mitochondria in ERAD-deficient cells, we labeled the ER with calnexin and the mitochondria with TOM20 and then quantified MAM changes by analyzing the degree to which calnexin and TOM20 colocalized. As shown in Fig. 4D-4H, the amount of MAM in Sel1L−/− cells was significantly (p < 0.05) higher than that in WT cells. Correlative studies have demonstrated that an increased number of MAM promotes higher mitochondrial Ca2+ levels (Arruda et al. 2014). As a result, ERAD deficiency may lead to an increase in mitochondrial Ca2+ concentration by altering the number of MAM in hepatic cells.
3.4. Reduced MAM Ca2+ partially restores mitochondrial bioenergetics in ERAD deficient cells
The effect of an imbalance in mitochondrial Ca2+ homeostasis on mitochondrial respiration is widely recognized. Given the finding that ERAD deficiency caused a substantial influx of ER Ca2+ into mitochondria, leading to an increase in mitochondrial Ca2+, we tested whether altered mitochondrial Ca2+ levels contribute to defective mitochondrial bioenergetics in ERAD-deficient liver cells. To decrease ER Ca2+ influx into mitochondria, we introduced 2APB into HepG2 cells. 2APB partially reversed the EerI-induced reduction in ATP levels (Fig. 5A). IP3R is an ER Ca2+ release receptor; IP3R siRNA was transfected into WT and SEl1L−/− cells, and the knockdown effect is shown in Fig. 5B. In Sel1L−/− cells, we found that the knockdown of IP3R expression partially restored MMP (Fig. 5C) and ATP levels (Fig. 5D).
Given the finding that ERAD deletion may boost ER Ca2+ influx into the mitochondria by increasing the number of MAM (Fig. 4D-4H), we used an outer mitochondrial membrane (OMM) linker plasmid that decreased the number of MAM (Fig. S2B). We found that OMM linker expression reduced mitochondrial Ca2+ levels (Fig. 5E) and improved OCR (Fig. 5F) in Sel1L−/− cells. Together, these findings show that ERAD deficiency compromises mitochondrial bioenergetics by increasing MAM and encouraging a substantial input of ER Ca2+ into mitochondria.
3.5. ERAD mediates palmitic acid-induced mitochondrial dysfunction in HepG2 cells
Palmitic acid (PA) is a significant contributor to the development of nonalcoholic fatty liver disease (NAFLD) and can induce lipid accumulation in the liver through its impact on mitochondrial function (Silva Figueiredo et al. 2017; Marra et al. 2018). A recent proteomic analysis revealed that treatment of HepG2 cells with PA damaged the protein quality control machinery (autophagy, ERAD, and the ubiquitin-proteasomal system) (Fig. 6A) (Saha et al. 2022). We tested whether ERAD mediated the impairment of mitochondrial function in hepatocytes caused by PA. We first performed cycloheximide (CHX)-based pulse-chase analysis for folding -deficient mutant NHK in HepG2 cells treated with and without PA. Folding-deficient mutant NHK showed efficient degradation in HepG2 cells without PA, but not in HepG2 cells treated with PA (Fig. 6B, C). Next, we investigated the effects of PA on MAM and mitochondrial Ca2+. As shown in Fig. 6D-6G, PA exposure increased MAM and mitochondrial Ca2+ levels in HepG2 cells. According to other studies, PA lowers the OCR (Mthembu et al. 2024). This study confirmed that PA impairs OCR and combining it with the ERAD inhibitor EerI further decreases OCR; yet the OMM linker restored the PA-induced OCR reduction (Fig. 6H). Together, these data suggest that PA exposure in HepG2 cells impairs ERAD function, and that ERAD is likely a mediator of PA impairment of mitochondrial function.
The ER and mitochondria interact both physically and functionally, allowing them to control each other's functions. A recent study revealed that ER protein quality control (ERAD) plays a crucial role in maintaining optimal levels of sigma receptor 1 (SigmaR1) to regulate mitochondrial dynamics in brown adipose cells (Zhou et al. 2020). In this study, we investigated whether the protein quality control system influences mitochondrial function in hepatic cells. Our research showed that ERAD facilitates Ca2+ signaling in the mitochondria by controlling the quantity of MAM. ERAD deficiency results in altered mitochondrial structure, reduced mitochondrial energy production, and increased Ca2+ transfer from the ER to the mitochondria by increasing the number of MAM. Knockdown of IP3R or a reduction in the number of MAM partially restores mitochondrial Ca2+ signaling and bioenergetics in ERAD-deficient hepatic cells. Moreover, we observed that PA disrupted ERAD protein degradation, leading to impaired mitochondrial function in hepatocytes. Collectively, these results suggest that ERAD is an important regulator of mitochondrial function, at least partially, by maintaining ER-mitochondrial Ca2+
homeostasis.
The physiological significance of ERAD in mitochondrial function in hepatic cells was first illustrated through in vitro experiments using the human hepatocellular carcinoma cell line HepG2. HepG2 cells treated with the ERAD inhibitor EerI showed a significant decrease in mitochondrial energy production and membrane potential (Fig. 1A, B and 1G, L). A similar reduction in mitochondrial energy production was observed in HepG2 cells with targeted disruption of the key ERAD protein, SEL1L (Fig. 2E). Overall, these data strongly suggested that ERAD plays a crucial role in facilitating mitochondrial bioenergetics in hepatic cells. To the best of our knowledge, this is the first direct experimental study linking ERAD deficiency to faulty mitochondrial bioenergetics.
The central focus of the present study was to clarify how ERAD deficiency mechanistically impairs mitochondrial function in hepatic cells. To this end, we first investigated whether changes in intracellular Ca2+ levels affect mitochondrial bioenergetics in ERAD-deficient hepatic cells. This investigation was prompted by the following: 1) Our previous study found that ERAD-defective β-cells exhibit aberrant intracellular Ca2+ levels; and 2) Numerous studies have demonstrated that intracellular Ca2+ is a critical signal mediating mitochondrial bioenergetics (Vecellio et al. 2024; Huo et al. 2024; Dridi et al. 2023). The present study found a significant increase in mitochondrial Ca2+ levels (Fig. 3A, B) and a decrease in ER Ca2+ (Fig. 3E, F) in ERAD-deficient HepG2 cells induced by SEL1L knockout. These SEL1L knockout-induced changes in ER and mitochondrial Ca2+ levels were prevented by the ER Ca2+ channel blocker 2-APB (Fig. 3G). More importantly, the negative impact of ERAD deficiency on mitochondrial bioenergetics was reversed by treatment with the ER Ca2+ channel inhibitor 2-APB and siRNA against IP3R (Fig. 5A, D). These results suggest that a deficiency in ERAD function hinders mitochondrial bioenergetics by increasing ER Ca2+ entry into the mitochondria.
Ca2+ fluorescent probes and immunofluorescence experiments further revealed that ERAD deficiency promotes ER Ca2+ entry into the mitochondria via MAM in hepatic cells. Analysis using a Ca2+ fluorescent probe demonstrated that the absence of SEL1L led to an increase in cytoplasmic Ca2+ in the basal state (Fig. 4A). However, the increase in cytoplasmic Ca2+ is similar in both WT and SEL1L−/− HepG2 cells after ATP stimulation (Fig. 4B). These findings suggest that ER Ca2+ is transported into the mitochondria through MAM in ERAD-deficient hepatic cells. Immunofluorescence analysis showed that SEL1L deficiency resulted in increasing the number of MAM in HepG2 cells (Fig. 4D–H). Increased amounts of MAM in hepatocytes under high-fat conditions promote the transfer of ER Ca2+ into mitochondria (Arruda et al. 2014). Although the precise molecular mechanisms underlying the increase in MAM numbers remain to be elucidated, the promotion of excessive ER Ca2+ entry into mitochondria Ca2+ is likely through an increase in MAM number in ERAD-deficient hepatic cells.
There is a link between mitochondrial dysfunction and many chronic metabolic diseases, such as type 2 diabetes, fatty liver, and cardiovascular disease, and PA plays an important role in this process (Ly et al. 2017; Liu et al. 2023; Riccardi et al. 2004). Recent studies have shown that PA can disrupt protein quality control machinery and mitochondrial function in liver cells. Importantly, our study demonstrated that the reduction in mitochondrial oxidative phosphorylation caused by PA was magnified in the absence of ERAD, and this effect could be blocked by a reduction in the number of MAM (Fig. 6H). Furthermore, we found that PA disrupted the function of ERAD, increased MAM number, and increased mitochondrial Ca2+ levels in hepatic cells (Fig. 6B-G). These findings suggested that the absence of ERAD exacerbated the effects of PA on mitochondrial dysfunction. Although it is necessary to elucidate the precise molecular mechanisms underlying the observed synergy between PA and ERAD deficiency in mitochondrial dysfunction, ERAD is likely to play an important role in PA-induced mitochondrial dysfunction by affecting MAM Ca2+.
In conclusion, our findings from pharmacological and genetic experiments highlight the critical role of ERAD in controlling mitochondrial function in human hepatic cells. Mechanistically, ERAD deficiency induces functional damage to mitochondria by impairing MAM Ca2+ homeostasis. Additionally, ERAD mediates PA-induced mitochondrial damage in liver cells. This study offers novel insights into the interactions between the ER and mitochondria and has important implications for understanding the molecular mechanisms of mitochondrial dysfunction-related diseases such as NAFLD.
Acknowledgments
We thank Yuanyuan Gao, Qiaocheng Zhai, and Lin Zhang for technical assistance; QiWu, and Lu Guo for expression plasmids or other reagents. Dr. Feng Wang, Qiaoming Long for helpful discussions and critical comments on the manuscript.
Author Contributions
Y.H. generated Sel1L-deficient HepG2 cell lines, performed the vast majority of in vitro experiments, drafted the initial version. Y.B. performed Immunofluorescence experiments. J.B and M.W provided key reagents and helpful discussions. F.Z. designed the experiments and wrote the manuscript. All authors reviewed and approved the manuscript. F.Z. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This work was supported by National Natural Science Foundation of China grant 82100893.
Data availability
The article and supplementary material contain the data that back up the findings of this study. The corresponding author will provide access to all relevant raw data upon reasonable request.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Consent for publication
All authors approved this manuscript for publication.
- Arora A, Taskinen JH, Olkkonen VM. Coordination of inter-organelle communication and lipid fluxes by OSBP-related proteins. Prog Lipid Res. 2022;86:101146.
- Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–35.
- Baillat, D., Russell, W. K., and Wagner, E. J. (2016) CRISPR-Cas9 mediated genetic engineering for the purification of the endogenous integrator complex from mammalian cells. Protein Expr. Purif. 128, 101–108.
- Beaulant A, Dia M, Pillot B, Chauvin MA, Ji-Cao J, Durand C, Bendridi N, Chanon S, Vieille-Marchiset A, Da Silva CC, Patouraux S, Anty R, Iannelli A, Tran A, Gual P, Vidal H, Gomez L, Paillard M, Rieusset J. Endoplasmic reticulum-mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J Hepatol. 2022;77(3):710–722.
- Christianson JC, Jarosch E, Sommer T. Mechanisms of substrate processing during ER-associated protein degradation. Nat Rev Mol Cell Biol. 2023;24(11):777–796.
- Dridi H, Santulli G, Bahlouli L, Miotto MC, Weninger G, Marks AR. Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart. Biomolecules. 2023;13(9):1409.
- Elwakiel A, Mathew A, Isermann B. The role of endoplasmic Reticulum - mitochondria -associated membranes in diabetic kidney disease. Cardiovasc Res. 2024;119 (18): 2875–2883.
- Henne WM. Organelle homeostasis principles: How organelle quality control and inter-organelle crosstalk promote cell survival. Dev Cell. 2021;56(7):878–880.
- Hu Y, Gao Y, Zhang M, Deng KY, Singh R, Tian Q, Gong Y, Pan Z, Liu Q, Boisclair YR, Long Q. Endoplasmic Reticulum-Associated Degradation (ERAD) Has a Critical Role in Supporting Glucose-Stimulated Insulin Secretion in Pancreatic β-Cells. Diabetes. 2019;68(4):733–746.
- Huo J, Molkentin JD. MCU genetically altered mice suggest how mitochondrial Ca2 + regulates metabolism. Trends Endocrinol Metab. 2024 Apr 29:S1043-2760(24)00088 – 2.
- Jang W, Puchkov D, Samsó P, Liang Y, Nadler-Holly M, Sigrist SJ, Kintscher U, Liu F, Mamchaoui K, Mouly V, Haucke V. Endosomal lipid signaling reshapes the endoplasmic reticulum to control mitochondrial function. Science. 2022;378(6625):eabq5209.
- Kang JA, Jeon YJ. How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases. Int J Mol Sci. 2021;22(4):2078.
- Liu J, Yang J. Mitochondria-associated membranes: A hub for neurodegenerative diseases. Biomed Pharmacother. 2022;149:112890.
- Liu Q, Yang X, Long G, Hu Y, Gu Z, Boisclair YR, Long Q. ERAD deficiency promotes mitochondrial dysfunction and transcriptional rewiring in human hepatic cells. J Biol Chem. 2020;295(49):16743–16753.
- Liu W, Zhu M, Gong M, Zheng W, Zeng X, Zheng Q, Li X, Fu F, Chen Y, Cheng J, Rao Z, Lu Y, Chen Y. Comparison of the Effects of Monounsaturated Fatty Acids and Polyunsaturated Fatty Acids on Liver Lipid Disorders in Obese Mice. Nutrients. 2023;15(14):3200.
- Ly LD, Xu S, Choi SK, Ha CM, Thoudam T, Cha SK, Wiederkehr A, Wollheim CB, Lee IK, Park KS. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. 2017;49(2):e291.
- Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68(2):280–295.
- Mthembu SXH, Mazibuko-Mbeje SE, Silvestri S, Orlando P, Marcheggiani F, Cirilli I, Nkambule BB, Muller CJF, Tiano L, Dludla PV. Low levels and partial exposure to palmitic acid improves mitochondrial function and the oxidative status of cultured cardiomyoblasts. Toxicol Rep. 2024;12:234–243.
- Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17(2):69–82.
- Riccardi G, Giacco R, Rivellese AA. Dietary fat, insulin sensitivity and the metabolic syndrome. Clin Nutr. 2004;23(4):447–56.
- Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13(10):607–25.
- Saha S, Verma R, Kumar C, Kumar B, Dey AK, Surjit M, Mylavarapu SVS, Maiti TK. Proteomic analysis reveals USP7 as a novel regulator of palmitic acid-induced hepatocellular carcinoma cell death. Cell Death Dis. 2022;13(6):563.
- Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112(36):11389–94.
- Sha H, He Y, Chen H, et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab 2009;9:556–564.
- Silva Figueiredo P, Carla Inada A, Marcelino G, Maiara Lopes Cardozo C, de Cássia Freitas K, de Cássia Avellaneda Guimarães R, Pereira de Castro A, Aragão do Nascimento V, Aiko Hiane P. Fatty Acids Consumption: The Role Metabolic Aspects Involved in Obesity and Its Associated Disorders. Nutrients. 2017;9(10):1158.
- Sun XN, An YA, Paschoal VA, de Souza CO, Wang MY, Vishvanath L, Bueno LM, Cobb AS, Nieto Carrion JA, Ibe ME, Li C, Kidd HA, Chen S, Li W, Gupta RK, Oh DY. GPR84-mediated signal transduction affects metabolic function by promoting brown adipocyte activity. J Clin Invest. 2023;133(24):e168992.
- Vecellio Reane D, Serna JDC, Raffaello A. Unravelling the complexity of the mitochondrial Ca2 + uniporter: regulation, tissue specificity, and physiological implications. Cell Calcium. 2024;121:102907.
- Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 2018;361(6401):eaan5835.
- Zhou Z, Torres M, Sha H, Halbrook CJ, Van den Bergh F, Reinert RB, Yamada T, Wang S, Luo Y, Hunter AH, Wang C, Sanderson TH, Liu M, Taylor A, Sesaki H, Lyssiotis CA, Wu J, Kersten S, Beard DA, Qi L. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science. 2020;368(6486):54–60.
No competing interests reported.
- floatimage8.png
Fig. S1 Fluorescence Mitochondrial morphology A-F, Fluorescent images of Sel1L+/+, Sel1L-/-, and EerI-treated Sel1L+/+ cells after stainning with mito-Tracker Green . Inset at the bottom right of each panel represents a magnified view of the dash line marked area.
- floatimage9.png
Fig. S2 Mitochondrial Ca2+ and MAM quantity assays A, detection of mitochondrial Ca2+ in HepG2 cells treated with EerI. B, detection of ER-Mitochondrial co-localization after OMM linker expression in cells.
{kind=link}
{kind=link}