Multiplex tissue imaging and analysis
The procedure is similar to a previously described method [23]. Briefly, a formalin-fixed, paraffin-embedded block was sliced into 3 µm-thick sections, and a tissue section was mounted on a glass coverslip, deparaffinized by heating at 60°C for 20 min, and then submerging in xylene. The tissue was then rehydrated in a graded ethanol series. The sections were immersed in citrate buffer solution (10 mm, pH 6.0) and heated in a pressure cooker for antigen retrieval. After cooling to room temperature, the tissue was stained with antibodies for 3 hours at room temperature in a humidity chamber. After fixation with a post-staining fixing solution for 10 min, 100% methanol for 5 min, and final fixative solution for 20 min, the tissue was stored in PBS at 4 ℃ until image acquisition. Multiplex imaging was executed using a microfluidics instrument (Akoya Biosciences, Menlo Park, CA), a fluorescence microscope (BZ-X700; Keyence, Osaka, Japan), and CODEX Instrument Manager software (Akoya Biosciences). An automated microfluidics system performed iterative annealing and stripping of fluorophore-labeled oligonucleotide barcodes complementary to the barcodes attached to antibodies. The raw image files were processed using CODEX processor version 1.8 (Akoya Biosciences). The antibodies and fluorophore-labeled oligonucleotide barcodes are described in Table S1.
QPTIFF files were imported to QuPath version 0.3.2. We extracted six 500 µm × 500 µm square regions in the image and performed cell segmentation on the basis of the nuclear stain using the cell detection algorithm in the software. To classify cells as being positive or negative for each marker, we generated training data for cell classification by deciding the positive and negative categories for every 10–30 cells in the regions based on fluorescence intensity. The results comprising the position (X,Y) and fluorescence intensity of each cell were exported as a csv file.
The csv file was imported to CytoMAP version 1.4.21. A detailed description of the workflow and functions built into CytoMAP is available in the online user manual. The representative area was subdivided into four regions by a 50 µm-radius raster-scanned neighborhoods function, which uses the Davies Bouldin Score and self-organizing map clustering methods, based on each fluorescence intensity. The analyses for neighborhood composition and Pearson correlation coefficients were performed based on the fluorescence intensity using the “Region Heatmaps” and “Cell-Cell Correlation” functions.
Preparation of fusion proteins
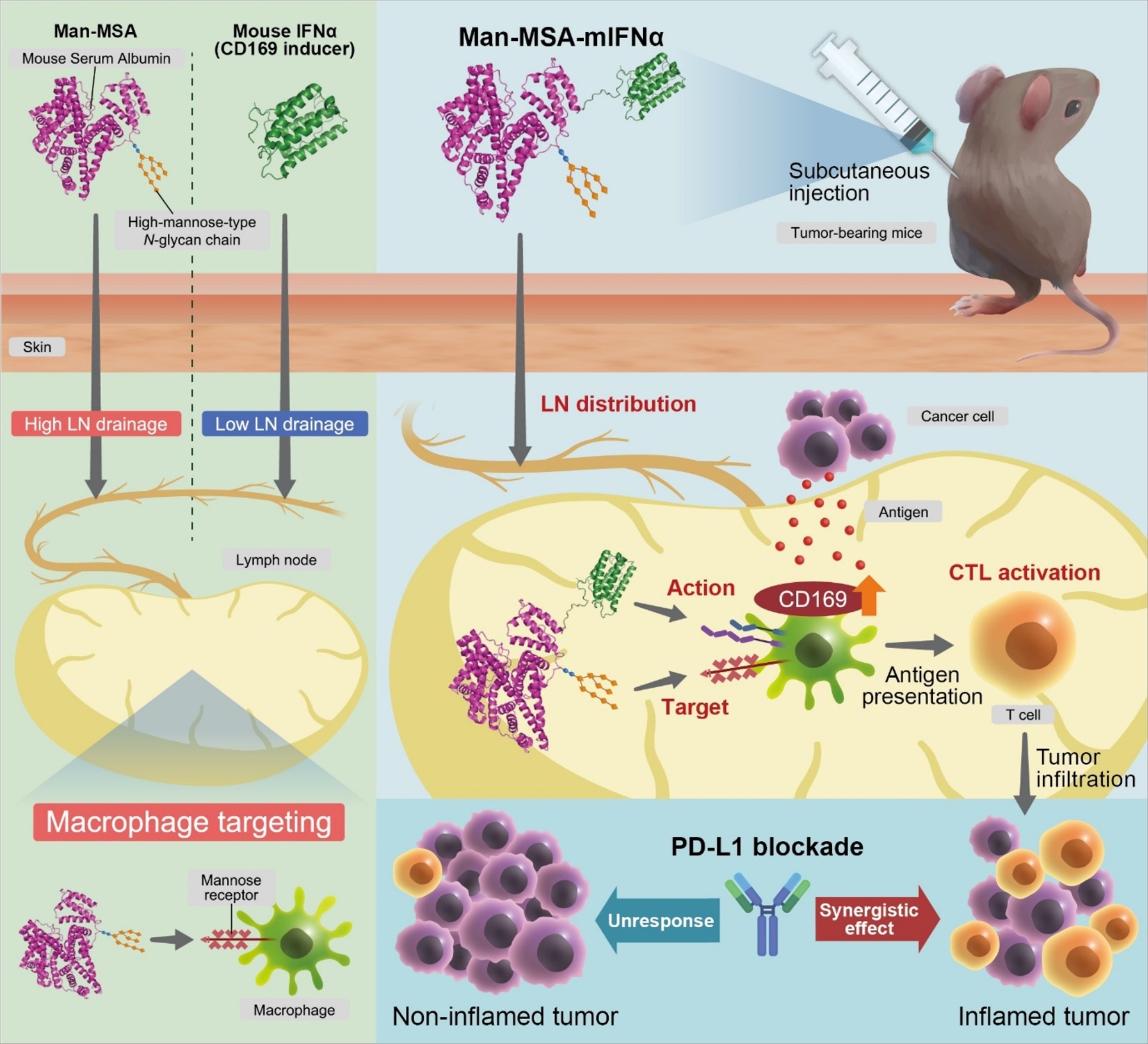
The designed fusion proteins are composed of MSA or MSA (D494N) linked to mIFNα2 (N78Q) via a polypeptide linker (-(Gly-Gly-Gly-Gly-Ser)2-). In this study, we replaced asparagine at position 78 of mIFNα2 with glutamine (N78Q) to remove a glycosylation sequence originally present in mIFNα2. The flow chart describing the creation of the MSA (D494N)-mIFNα2 (N78Q) gene using pPIC9 is shown in Fig. S1. PCR was performed using a PfuTurbo DNA polymerase. To isolate the DNA fragment of the base sequence coding for MSA, restriction enzyme Xho1 and Ava1 recognition regions were inserted into the 5′ and 3′ terminals, respectively. Similarly, restriction enzyme Ava1 and EcoR1 recognition regions were inserted into the 5′ and 3′ terminals of the DNA sequence encoding mIFNα2. Then, pPIC9 was digested with Xho1 and EcoR1, and the appropriate pPIC9 fragment was extracted by agarose gel electrophoresis. To obtain the cDNA construct coding for MSA-mIFNα2, DNA fragments (pPIC9, MSA and mIFNα2) were ligated overnight at 16°C using a DNA ligation kit (Takara bio, Shiga, Japan). The subsequent mutation of MSA (D494N) and mIFNα2 (N78Q) was performed using a Quick Change kit (Takara bio) with the mutagenic primers described in Table S2. Hexa-His-tag was inserted into the C-terminal of mIFNα2 (N78Q), and Pichia pastoris (SMD1168 strain) was transformed with pPIC9-MSA (D494N)-mIFNα2 (N78Q) by electroporation according to the manual.
Pharmacokinetic analysis of 125I-labeled proteins
The proteins were radiolabeled with 125I according to previously reported procedures [24]. 125I-labeled Man-MSA-mIFNα and MSA-mIFNα (1 mg protein kg− 1; approximately 7 × 106 counts per minute [CPM] per mouse), MSA (1 mg protein kg− 1; approximately 6 × 106 CPM per mouse), and mIFNα (1 µg protein kg− 1; approximately 8 × 105 CPM per mouse) were subcutaneously administered into the inguinal region of ICR mice. Inguinal LNs were isolated at each time point after the administration of the 125I-labeled proteins, and the radioactivity was measured using an autowell gamma counter (AccuFlex γ7001; Hitachi Aloka Medical, Tokyo, Japan) according to the manufacturer's instructions.
Preparation of Cy5-labeled proteins
The lyophilized proteins were dissolved in deionized water to 20 mg mL− 1. Cy5 NHS ester (0.5 mg) was added to the protein solution (1 mL), and the solution was mixed by rotation for 1 hour at room temperature. The unreacted Cy5 NHS ester was removed by ultrafiltration using an Amicon Ultra Ultracel-PL 10 kDa membrane filter (Merck Millipore, Burlington, MA).
Pharmacokinetic analysis of Cy5-labeled proteins
To evaluate the LN macrophage-targeting ability of Man-MSA-mIFNα, Cy5-labeled fusion proteins (60 nmol kg− 1) were subcutaneously administered into the inguinal region of ICR mice, and the inguinal LNs were isolated 1 hour after administration.
Flow cytometry: LNs or tumors were harvested, minced finely with scissors on ice, and then incubated with collagenase D (Sigma-Aldrich) or Dri tumor & tissue dissociation reagent (Becton Dickinson, NJ) at 37°C for 30 min. After passing the digested mixture through a 70 µm filter, the cells were incubated with ACK lysis buffer (NH₄Cl [0.15 m], KHCO₃ [10 mm], and EDTA [0.1 mm]) to remove red blood cells and suspended in FACS buffer (EDTA/PBS [2 mm] containing 0.5% FBS). The cells were incubated with FcR blocking reagent (1:50, Miltenyi Biotec, 130-092-575) and Fixable Viability Dye eFluor 780 (1:5,000, Invitrogen, 65-0865-14) at room temperature for 5 min and then, stained with antibodies for cell surface markers at 4°C for 15 min. After washing with FACS buffer, the cells were fixed, permeabilized, and incubated with antibodies for intracellular markers at 4°C for 30 min. The cells were analyzed on a FACSVerse (Becton Dickinson) flow cytometer with FACSuite (Becton Dickinson) software. The data were analyzed using FlowJo software (Becton Dickinson). The gating strategies are shown in Fig. S2 and Fig. S3, and the antibodies in Table S1.
Evaluation of CD169 expression in vitro
J774.1 cells were seeded at a density of 3.0 × 105 cells per well in 12-well plates and incubated overnight at 37°C before the addition of mIFNα (1 IU mL− 1), MSA, MSA-mIFNα, or Man-MSA-mIFNα (70 nm) to the cells. The cells were collected after incubation for 24 hours at 37°C, and CD169 expression was evaluated by flow cytometry.
Evaluation of CD169 expression in vivo
We first assessed the optimal dose of Man-MSA-mIFNα and the time course of CD169 expression. As shown in Fig. S4 and Fig. S5, we confirmed that, 72 hours after administration, Man-MSA-mIFNα (0.6 nmol kg− 1) showed high CD169 expression on LN macrophages. Next, to compare the in vivo CD169 induction levels of mIFNα, MSA-mIFNα and Man-MSA-mIFNα, we evaluated the titer of mIFNα using J774.1 cells and found the induction level of CD169 after treatment with mIFNα (1 IU mL− 1) to be equivalent to that after Man-MSA-mIFNα (6 µg mL− 1, 0.07 nmol mL− 1) (Fig. S6). Thus, mIFNα (8 IU kg− 1), MSA-mIFNα (0.6 nmol kg− 1), or Man-MSA-mIFNα (0.6 nmol kg− 1) was subcutaneously administered into the inguinal region of C57BL/6N mice. Seventy-two hours after administration, inguinal LNs were harvested, and the CD169 expression on macrophages evaluated by flow cytometry.
Antitumor effects of Man-MSA-mIFNα
We first evaluated the optimal dosing interval using MB49-bearing mice and found that the administration of Man-MSA-mIFNα once every 4 days, but not once every 7 days, exerted significant antitumor effects (Fig. S7). Therefore, mIFNα (8 IU kg− 1), MSA-mIFNα, and Man-MSA-mIFNα (0.6 nmol kg− 1) were injected subcutaneously near the tumor once every 4 days. For combination therapy, the mice were treated intraperitoneally with isotype control antibodies (200 µg; BioLegend, 400667) and anti-PD-L1 antibodies (200 µg; BioLegend, 124329) on the 10th and 13th days after tumor inoculation and then, subcutaneously with Man-MSA-mIFNα (0.3 nmol kg− 1) once every 4 days. The tumor volume was calculated according to the formula V = 0.4 × a × b2, where “a” is the major axis, and “b” is the minor axis of the tumor. Tumor-bearing mice were sacrificed on the 18th day after tumor inoculation.
Statistical analysis
The experimental data were analyzed using GraphPad Prism 9. The means for two-group data were compared by using an unpaired t-test, and the means for more than two groups were compared by one-way ANOVA followed by Tukey’s multiple comparison or Bonferroni's multiple comparison. The probability value of p < 0.05 was considered to be a statistically significant difference.
{kind=link}