Preparation of GFAP immunomagnetic liposome beads (GFAP-IMLs)
GFAP-IMLs were prepared by using the reverse-phase evaporation (REV) method. Briefly, 5 mg dioleoylphosphocholine (DOPC) (Avanti Company) and 5 mg cholesterol were obtained and added into a 50 ml 3-neck flask. After removing ethanol, 1.0 mg Fe3O4-hydrophobic magnetic nanoparticles (HMNs) were dissolved in 3.0 ml CH2Cl2 and transferred into the previously prepared 3-neck flask. The mixture in the flask was sonicated on ice. Simultaneously, 2mg GFAP antibody (Abcam, ab7260) modified glycidyl hexadecyl dimethylammonium chloride (GFAP-GHDC) was dissolved in 6 ml ddH2O and gradually added to this flask. The preparation method of GFAP-GHDC refers to our previous method [36]. After emulsification, rotary evaporation was used to remove the residual CH2Cl2 from the emulsion. The solution was magnetically separated and washed three times, and GFAP-IMLs were obtained. Similarly, EpCAM (Abcam, ab71916) modified IMLs were constructed. The detailed preparation process and reagent consumption are described in our previous study [37].
Characterization of IML
Zetasizer Nano-ZS 90 (Malvern Instruments, Ltd., UK) was used to determine the particle size and potential of IMLs. Atomic force microscopy (AFM) was used for the micromorphology of different IMLs. Magnetic hysteresis loops of these magnetic particles were detected using PPMS-9 (QUANTUM DESIGN, USA). An ultraviolet spectrophotometer was used to confirm the presence of antibodies on the surface of IMLs and to analyze the antibody content qualitatively. A bicinchoninic acid assay (BCA assay) was used to quantitatively analyze the antibody content of these IMLs. Polyacrylamide gel electrophoresis (PAGE) was used to detect the antibody content and to confirm the presence of the antibodies on the surface of these IMLs.
Cytotoxicity Assay of the IMLs
U87, U251 and D425 Cells were suspended and seeded into a 96-well plate with 8,000 cells in each well containing 100 μl culture medium and cultured overnight. IMLs were added into each well at a final concentration gradient of 0, 10, 50, 100, 200, 500, and 1000 µg/ml. After culturing at 37°C for 48 hrs, 10 μl of 5 mg/ml MTT reagent was added to each well. After 3 hrs of incubation, 150 μl DMSO was added to each well to dissolve the crystal after discarding the medium, followed by measuring at 490 nm using a spectrometer (SpectraMax M5/M5e).
Evaluation of the interaction between IMLs and brain tumor cells
Commercial glioma cells U87, U251 and D425 were routinely cultured in DMEM complete culture medium supplemented with 10% FBS and 1% penicillin-streptomycin in a humidified 5% CO2 incubator at 37°C. The cytotoxicity assay of IMLs is described in the supplementary file. Sterile slides were placed in a 24-well plate, and 1×104 U87 cells were seeded into each well containing 1 ml culture medium. These cells were cultured in a 5% CO2 incubator at 37°C for 24 hrs. After refreshment with new medium, IMLs were added to each well at 20 μl/well, and an equal volume of PBS was added to the control wells. After 48 hrs of incubation, the medium in each well was discarded and washed with PBS. After fixed the cell with paraformaldehyde, 100 μl DAPI staining solution was added to each well and incubated for 5 minutes. After discarding the staining solution and washing with PBS, cell slides were removed from the wells and placed in an inverted position on a slide-proof glass slide coated with antifade mountant. These slides were observed by a confocal laser scanning microscope (Lcica TCS SP8 STED 3X).
Rhodamine 123 (R123) (Sigma) was encapsulated in the GFAP-IMLs. After dialysis for 5 hrs to remove the excess free R123, R123-labeled GFAP-IMLs were mixed with U87-GFP cells and incubated at 37°C for different minutes. The final dyeing effect of U87-GFP cells was observed under a fluorescence microscope after washing with PBS.
Experiment on the recovery rate of simulated CTCs by IML
Different (10-200) suspended U87 cells were mixed with 7.5 ml PBS with anticoagulant to mimic the CTCs in CSF and blood. The abilities of GFAP-IMLs, EpCAM-IMLs, EGFR-IMLs and IMLs to capture CTCs were measured. The cell suspension was dropped onto a slide-proof polylysine-coated glass slide, which was subjected to a fluorescence microscope after drying out. The experimental procedure of CTCs detection is the same as that in our previous study [36] and described in the following of the experimental procedure of CTCs detection.
U87-GFP cell separation efficiency of GFAP-IML in vitro and in vivo
U87-GFP cells at the logarithmic growth stage were digested with trypsin to make the cell suspension. A total of 0.2 ml of the prepared U87-GFP cell suspension (1×107/ml) was subcutaneously injected into the right back skin of 4-week-old female BALB/c nude mice (Supplied by SLRC Laboratory Animal). Tumor volume was calculated according to the following formula: Tumor volume V(mm3) = π/6×Length (mm)×Width (mm2). Measurement was performed once every 2 or 3 days, and the growth rate was calculated according to the following formula: Growth rate(%) = Mean tumor volume(mm3)/ Survival time of tumor-Bearing mice (days).
Blood samples (0.1 ml) were collected from nude mice eyeball, from which CTCs were isolated and identified by IML incubation followed by magnetic separation. Similar to the above CTC recovery method, the supernatant was transferred carefully into a 1.5 ml centrifuge tube, and an equal volume of PBS was added and mixed. Then, 30 μl IMLs was added and incubated at RT for 25 minutes and mixed once every 5 minutes. Furthermore, 30 μl DAPI, 10 μl EGFR-FITC (Abcam, ab11400) solution, and 10 μl CD45-PE were used to stain isolated cells for 15 minutes away from light. After washing three times with PBS, the cell suspension was dropped onto a slide-proof glass slide and observed under a fluorescence microscope.
The experimental procedure of CTCs detection
Each cell sample to be tested was centrifuged at 1000 rpm for 10 minutes. After centrifugation, the upper layer was carefully removed, leaving the lower layer solution in the tube, and equal volume PBS was added to the tube and mixed well. Then, 20 µl IMLs was added to each tube and incubated for 25 minutes at room temperature (RT) and mixed every 5 minutes. The tube was placed on a magnetic separation rack for 10 minutes, and the IMLs with captured CTCs were washed with PBS twice. DAPI and fluorescent antibody were added and incubated for 15 minutes away from light to stain the cells, followed by magnetic separation for 5 minutes at the end of staining. The cells were washed twice with deionized water to fully remove the unbound antibody and DAPI. Finally, 15 µl deionized water was added to the tube to resuspend CTCs. The cell suspension was dropped onto a slide-proof polylysine-coated glass slide for fluorescence microscope inspection.
Inclusion criteria for patients with brain tumor
The present study was approved by the hospital ethical committee and signed informed consents were obtained from all patients. (1) Children (age≤13) histologically diagnosed with brain tumor by two board certificated pathologists independently; (2) no metastatic lesions with diameter ≥ 1 cm by intracranial MRI; (3) no craniocerebral injury, brain surgery or radiotherapy history within the last 6 months; (4) the symptoms of intracranial hypertension was controlled by dehydration medication; (5) patients could tolerate the lumbar puncture for CSF collection; and (6) patients with intracranial meningiomas, meningioma and meningeal lesions were excluded from the study. 24 patients who met these criteria were enrolled in our study, with 12 males and 12 females.
Inclusion criteria for patients with non-tumor brain diseases
(1) Patients with non-tumor brain lesions, craniostenosis and scalp mass who were hospitalized in the same period as the experimental group; and (2) patients who needed external ventricular drainage, ventriculoperitoneal shunt, lumbar cistern drainage, lumbar puncture for CSF examination during hospitalization. 3 patients with communicating hydrocephalus met these criteria were included in the control group.
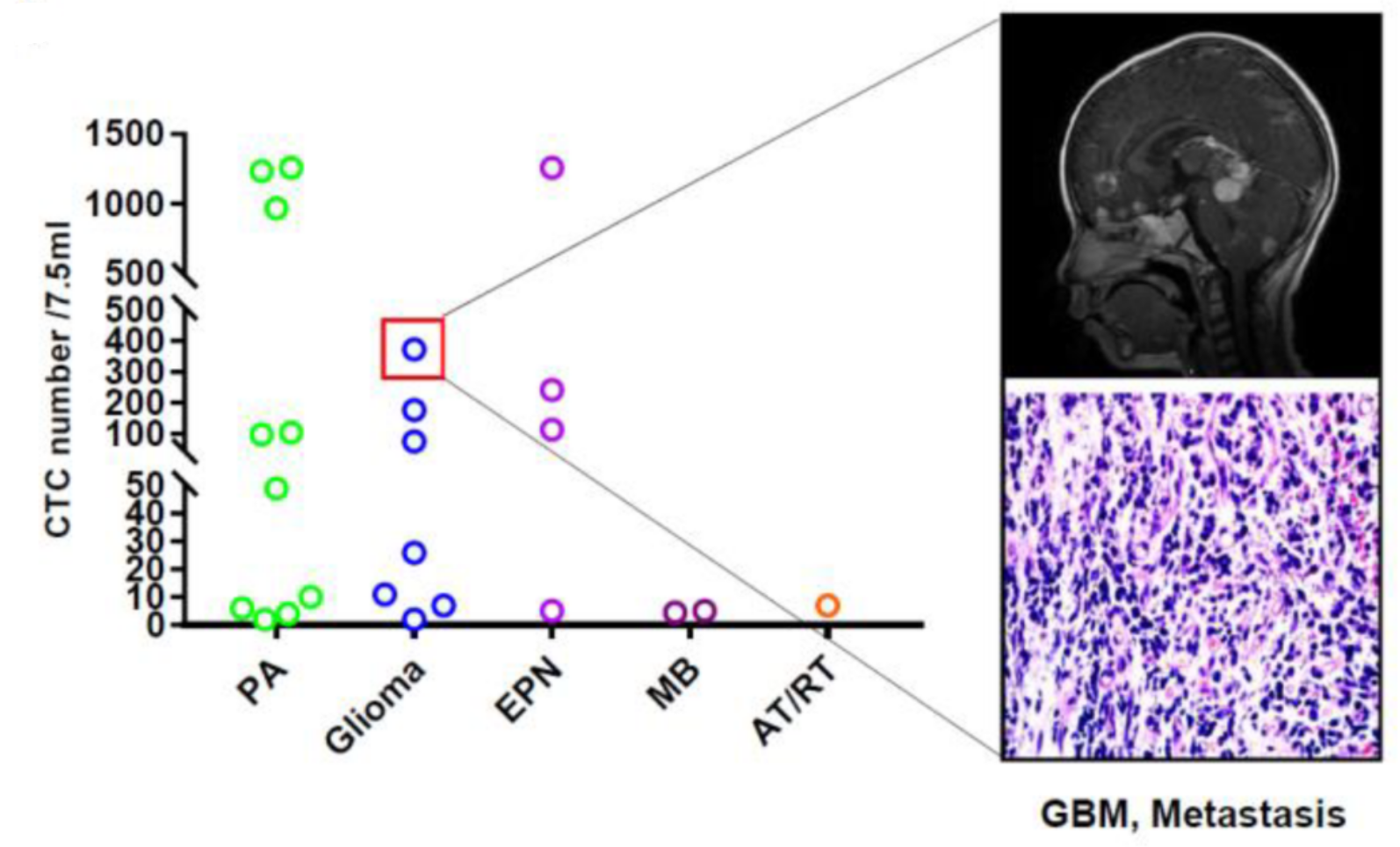
Detection of CTCs in tumor patients
This study was approved by the Ethics Committee of Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, and all patients involved in this study signed informed consent. For each patient before tumor resection, 7.5 ml of CSF through lumbar puncture and peripheral blood were collected in an EDTA anticoagulant tube for immediate laboratory examination. Self-prepared anti-GFAP, EpCAM and EGFR IMLs were used to enrich and screen the CTCs in CSF and peripheral blood samples. DAPI was used to identify the intact cells with nuclei, and then cells were stained with fluorescently labeled anti-CD45 and EGFR-FITC (for GFAP-IML)/GFAP-FITC (for EpCAM/EGFR-IML separation) monoclonal antibodies to distinguish the neuroepithelial tumor cells from the white blood cells. CTCs that met the evaluation criteria, i.e., GFAP+, EGFR+, DAPI+ and CD45-, were counted by using a complementary polychromatic fluorescence cell counting instrument.
DNA extraction and gene detection in CTCs
The CTCs enriched by GFAP-IMLs were used to extract DNA, which was performed according to the instructions of the DNA extraction kit (EZ Bioscience, No. B0007). PCR-related amplification reagents were purchased from Yeasen Biotech (Shanghai) Co., and Ltd. PCR primers were purchased from Sangon Biotech (Shanghai) Co., Ltd. H3F3A, HIST1H3B and HIST1H3C, as well as NPR3 primers were described in our previous literature[30].
H3F3A, HIST1H3B and HIST1H3C, as well as NPR3 primers are as follows:
CATGGCTCGTACAAAGCAGA (H3F3A-F) ;
GCAAAAAGTTTTCCTGTTATCCA(H3F3A-R);
TTGGGTCCAATAGTTGGTGGT(HIST1H3B-F);
GACTTTTGGTAGCGGCGGAT(HIST1H3B-R);
CGCGCGGGACTTTTGAAATA(HIST1H3C-F);
GCTCGGTGGACTTCTGGTAG(HIST1H3C-R);
CTACGCCTTCTTCAACATTGAG/TAGCTTCAAAGTCGTGTTTGTC(NPR3-F/R).
RNA sequencing steps are as follows. CTC isolated by GFAP-IMLs was collected into the RNase-free centrifuge tube, and RNA extraction kit was used for extraction. After quality verification, library construction and on-machine sequencing were carried out.
The obtained CTC-DNA could be sequenced by fluorescence quantitative PCR, sanger sequencing and whole exome sequencing (WES). The WES steps are as follows. 100 ng CTC-DNA from each sample was extracted for library construction, including pruning and ligation of aptamers. Agilent v2 Human Exon bait kit was used for exon group capture. Illumina HiSeq platform was used to sequence captured DNA samples and integrate paired sequencing readings for each sample.
Statistical analysis
Data from each group were processed by SPSS 19.0 statistical software and presented by Prism 7.0 software. Measurement data are presented as the mean ± SD and were compared by the rank sum test. A p value <0.05 was considered statistically significant. A heatmap was produced by R 3.3, and the number of CTCs was transferred by log transformation. For other methods employed in this manuscript, please refer to the supplementary information.
{kind=link}