Ethical statement
The informed consent was completed by each patient participating in this study before recording the information and sampling based on the Helsinki declaration. This study was approved by the Ethics Committee of Shahid Sadoughi University of Medical Sciences, Yazd, Iran.
Population study
This study included a population of patients (126 cases) with zoonotic CL caused by L. major that five of them had no response to Glucantime, which referred to the laboratory of Isfahan Diagnosis Center, Iran, from October 2017 to December 2019. The patients with L. major were followed for three months. The cases with no responses to Glucantime treatment were considered as TF isolates. The cases with the response to Glucantime were considered as treatment response (TR).
Sampling
Samples were collected by scrubbing the lesion edge after disinfecting by using 70% alcohol. Two slides were prepared for either direct microscopic examination or molecular analysis for each patient.
Detection and identification
A microscopic examination was carried out to find the Leishman body. The DNA was extracted from stained smears using the DNA extraction kit (GeneAll, South Korea) based on manufacturer instructions. The quantity of the isolated DNA was evaluated using a spectrophotometer by nanodrop (ABI, USA). With the aim of verification of Leishmania spp., ITS1-PCR was used using LITSr-F 5'-CTGGATCATTTTCCgATg-3' and L5.8s-R 5'-TGATACCACTTATCGCACTT-3' [12]. Each amplification reaction mixture included 1x PCR buffer, 0.2 mM dNTP, 1.5 mM MgCl2, 1.5 U Taq DNA polymerase, 0.5 µM each primer, and 100 ng DNA. The amplification temperature included the first denaturation 94 °C for 5 min that followed by 35 cycles of 94 °C for 45 s, 50°C for 45 s, and 72 °C for 45 s. The final elongation was done by 72 °C for 5 min. Positive and negative controls were considered in each run using L. major (MRHO/IR/75/ER) and ddH2O, respectively. The amplification analysis was done using agarose gel electrophoresis (1%) alongside with 50 bp DNA ladder. The expected amplicon was about 300-350 bp for verification of Leishmania spp. detection. Molecular identification was performed on positive amplicons digested using Hae III (Bsu RI) for 3 h at 37 °C. The digestion banding pattern analysis was assessed using 3% agarose gel electrophoresis alongside with 50 bp DNA ladder. L major (MRHO/IR/75/ER) was considered as positive control. A banding pattern revealing two main bands of 220 and 140 bp was considered as L. major. All tests were done in triplicate.
Molecular analysis of barcode gene of COXII
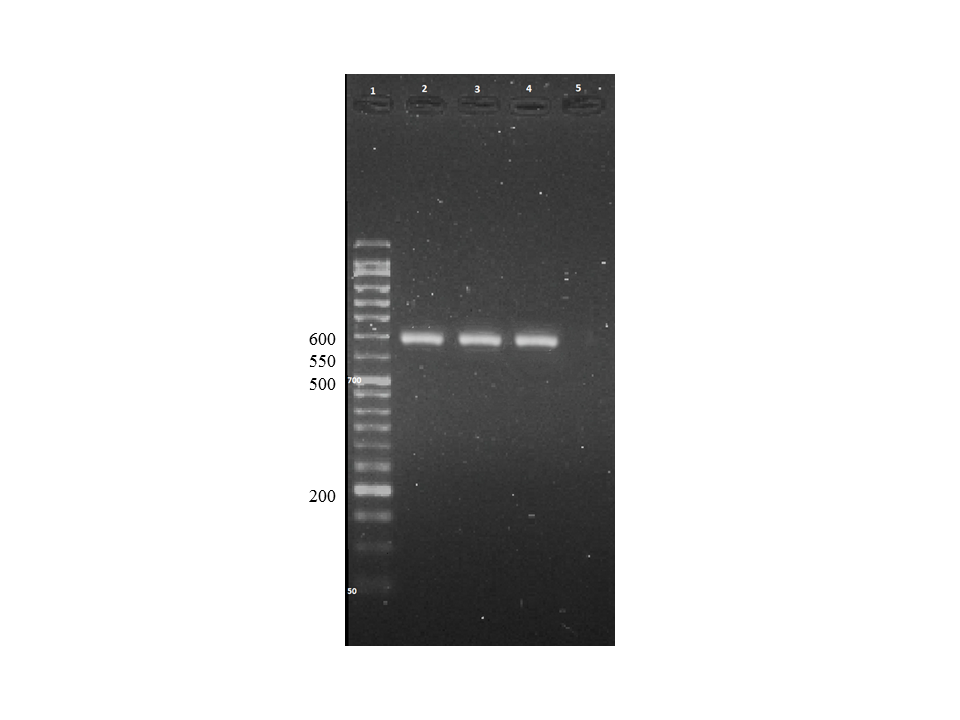
The primer pair of COXII was COXII-F 5'-GCATAAATCCATGTAAAACACCACA-3' and COXII-R 5'-TGGCTTTTATWTTATCATTTTGAATG-3' [13]. Each amplification reaction included 1x PCR buffer, 0.2 mM dNTP, 1.5 mM MgCl2, 1.5 U Taq DNA polymerase, 0.5 µM each primer, and 100 ng DNA. The amplification protocol was the first denaturation at 94 °C in 1 cycle, followed by 35 cycles of 94 °C for 60 s, 57 °C for 20 s, and 72 °C for 30 s. The final elongation was done by 72 °C for 5 min. Positive and negative controls were considered in each run with L. major (MRHO/IR/75/ER) and ddH2O, respectively. The amplification analysis was done using agarose gel electrophoresis (1.5%) alongside with 50 bp DNA ladder. The expected amplicon was 602 bp.
Molecular analysis of barcode gene of 13A/B
The primers of 13A/B and the reaction conditions were obtained from the study by Aghai-Maybodi et al. [14]. Positive and negative controls were considered in each run with L. major (MRHO/IR/75/ER) and ddH2O, respectively. The amplification analysis was done using agarose gel electrophoresis (3%) alongside with 50 bp DNA ladder. The expected amplicon was 120 bp.
Sequencing and molecular analysis
The amplicons of the target barcode gene of COXII in 21 cases, including 5 TF and 16 TR cases were purified and sequenced. The amplicons of the target gene of 13A/B regarding 5 TF isolates and 6 TR isolates were purified and sequenced. The sequences were repaired and BLAST. All the studied sequences were submitted in the GenBank database using BankIt (NCBI). The related sequences were searched from GenBank (NCBI) using BLASTn, https://blast.ncbi.nlm.nih.gov/Blast.cgi [15]. Multiple alignments were done using T-COFFE. The phylogenetic analysis was done using MEGA 7.0.21 [16]. The evolutionary history was inferred using the Neighbor-Joining method [17]. The evolutionary distances were computed using the Maximum Composite Likelihood method [18] and were in the units of the number of base substitutions per site. All cases were eliminated if they had gaps and missing data. Bootstrap of 1000 was performed to estimate the node reliability for COXII and 500 for 13A/B. Sequences of COXII were compared to seven entries retrieved from GenBank, including KU680818.1, KU680819.1, KF815210.1, KF815211.1, EU140338.1, AF287688.1, and EF633106.1. The sequences of 13A/B regarding to 5 TF and 6 TR isolates were compared each other and phylogenetic tree was drawn.
{kind=link}
{kind=link}
{kind=link}