The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been mutating since it was first sequenced in early January 2020. The genetic variants have developed into a few distinct clusters with different properties. Since the United States (US) has the highest number of viral infected patients globally, it is essential to understand the US SARS-CoV-2. Using genotyping, sequence-alignment, time-evolution, k-means clustering, protein-folding stability, algebraic topology, and network theory, we reveal that the US SARS-CoV-2 has four substrains and five top US SARS-CoV-2 mutations were first detected in China (2 cases), Singapore (2 cases), and the United Kingdom (1 case). The next three top US SARS-CoV-2 mutations were first detected in the US. These eight top mutations belong to two disconnected groups. The first group consisting of 5 concurrent mutations is prevailing, while the other group with three concurrent mutations gradually fades out. We identify that one of the top mutations, 27964C>T-(S24L) on ORF8, has an unusually strong gender dependence. Based on the analysis of all mutations on the spike protein, we further uncover that three of four US SASR-CoV-2 substrains become more infectious. Our study calls for effective viral control and containing strategies in the US.
Article
Characterizing SARS-CoV-2 mutations in the United States
https://doi.org/10.21203/rs.3.rs-49671/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 15 Feb, 2021
Version 1
posted
You are reading this latest preprint version




Supporting Tables spreadsheets are not available with this version.
There is NO Competing Interest.
- 2019nCoVEvolutionSupportingInformation.pdf
Supporting Information
- FigureS1.jpg
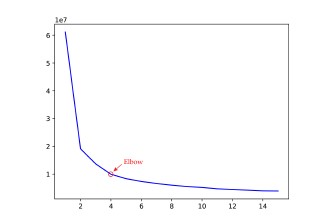
The plot of WCSS according to the number of clusters based on Jaccard distance metric. Here, Jaccard distance-based representation is taken as the input feature. The arrows point out the optimal number of clusters. The within-cluster sum of squares against the number of clusters for the SNP variants in the United States. The optimal number of clusters in the United States is four.
- FigureS2.jpg
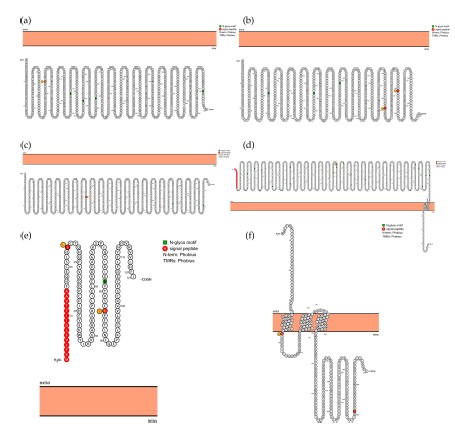
The visualization of six SARS-CoV-2 proteoforms. (a) Proteoform of NSP2. (b) Proteoform of NSP13. (c) Proteoform of NSP12. (d) Proteoform of spike protein. (e) Proteoform of ORF8. (f) Proteoform of ORF3a. The red color represents the wild type and the yellow represents the wild type.
{kind=link}
{kind=link}