Characterization of EVs of different origins
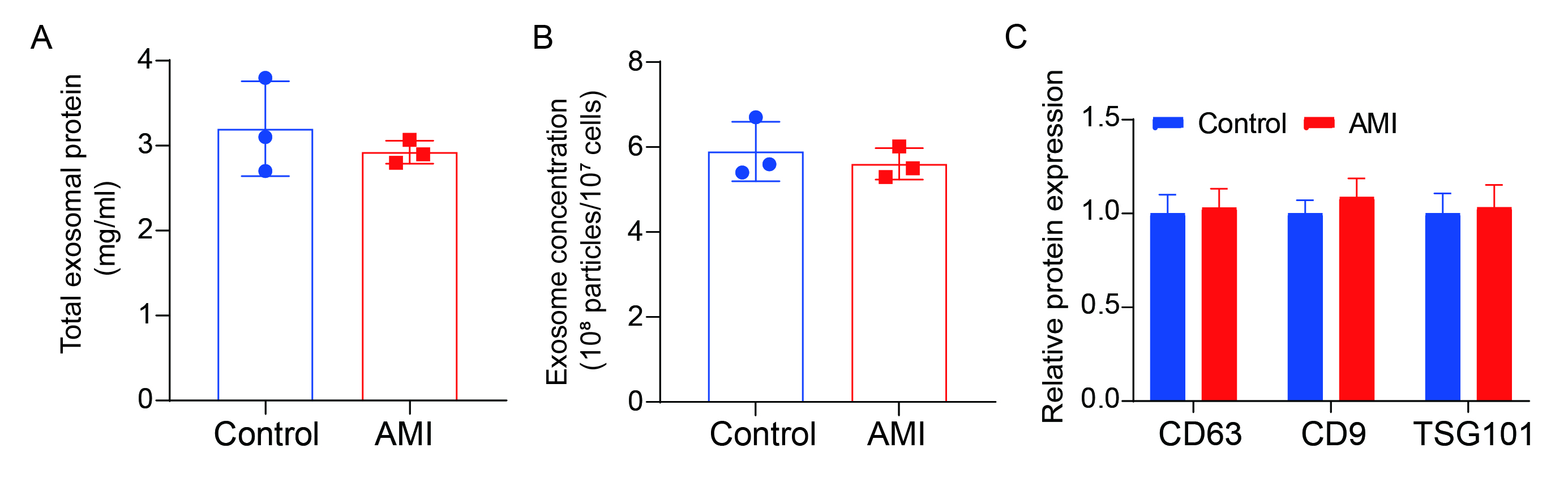
Firstly, circulating blood exosomes were extracted from both the control and AMI groups, and the concentration of exosomes and total protein was examined. The results showed no statistically significant difference in EVs between the two groups (Supplementary Fig. 1A-B). Additionally, WB detection of CD63, CD9 and TSG101 indicated that the purity and concentration of exosomes from both sources were similar (Supplementary Fig. 1C).
Regulatory effects of EVs from different sources on cardiomyocytes and endothelial cells
Initially, PKH67 was employed to label non-source EVs for validation of their ability to enter AC16 and HUVEC cells (Fig. 1A). Subsequently, the impact of co-culturing EVs from different sources on cell viability was assessed, revealing that N-EVs significantly enhanced cell proliferation. Conversely, AMI-EVs further suppressed cell proliferation as confirmed by EdU staining (Fig. 1B-C). Moreover, examination of apoptosis in both cell types demonstrated contrasting effects; AMI-EVs notably promoted apoptosis while N-EV inhibited it (Fig. 1D). Finally, evaluation of HUVECs' migration and tube-forming capacity revealed that N-EVs significantly augmented these functions whereas AMI-EVs hindered them (Fig. 1E-F).
Effects of EVs from different sources on cardiac function in a mouse model of acute myocardial infarction
We induced AMI in rats through coronary ligation, followed by intramyocardial injection of AMI-EVs, N-EVs, or PBS into the MI border zone. Left ventricular ejection fraction (LVEF) was utilized to evaluate cardiac function enhancement or maintenance. There were no significant differences in LVEF among the four treatment groups at baseline (Fig. 2A). Over the subsequent three weeks, the control group exhibited a gradual decrease in LVEF, while hearts injected with N-EVs demonstrated higher LVEF compared to those of the Sham or PBS groups. Conversely, AMI-EVs-treated hearts displayed worsened LVEF when compared to PBS-treated control hearts (Fig. 2B). In terms of myocardial injury treatment, we confirmed through histological staining that N-EVs could inhibit myocardial fibrosis; however, AMI-EVs aggravated these phenomena (Fig. 2C). Furthermore, based on our in vitro assays indicating that EVs from different sources could regulate angiogenesis, we assessed the expression of angiogenesis markers (VEGFA, CD34 and COX2) in myocardial tissue. Compared to the Sham and PBS groups, N-EVs treatment promoted angiogenesis while AMI-EVs inhibited their expression levels (Fig. 2D-E).
EVs confer protection against oxidative stress on cardiomyocytes and endothelial cells.
EVs play a crucial role in facilitating the exchange of materials and information between cells. To further validate the potential of EVs in mitigating H2O2-induced oxidative stress injury, HUVECs and AC16 (600 µmol/L) were subjected to H2O2 treatment and co-cultured with N-EVs. In CCK8 and EdU assays, N-EVs (100 µg/mL) exhibited protective effects on both HUVEC and AC16 against H2O2 after 24 hours (Fig. 3A-C). The cytoprotective effect of N-EVs against H2O2-induced injury was also confirmed through apoptosis assay (Fig. 3D). Moreover, in the tube formation assay, the total branch length observed for N-EVs-treated HUVECs surpassed that of PBS- or H2O2-treated counterparts, indicating that N-EVs effectively safeguard cardiomyocytes and endothelial cells from H2O2-induced injury (Fig. 3E-F).
The role of miR-133a-3p in EVs
Current research has demonstrated the pivotal role of miRNAs in orchestrating the regulatory mechanisms of EVs. Consequently, employing microarray analysis, we identified a total of 10 up-regulated miRNAs in N-EVs and observed significant down-regulation of 21 miRNAs (Fig. 4A-B). We selected the three most significantly up-regulated miRNAs (miR-133a-3p, miR-144-3p, and miR-23b-3p) for qRT-PCR validation and observed a significant increase in the expression of miR-133a-3p, suggesting its potential role as a crucial regulatory molecule in cardiomyocyte and endothelial cell proliferation mediated by N-EVs (Fig. 4C).
To further investigate the impact of miR-133a-3p on EV-mediated promotion of cardiomyocyte and endothelial cell proliferation, we treated AC16 and HUVEC cells with PBS, N-EV, or AMI-EV. Subsequent qRT-PCR analysis revealed a significant upregulation of miR-133a-3p expression specifically in the N-EV-treated group, while no notable changes were observed in cells treated with PBS or AMI-EV (Fig. 4D). Importantly, when we subjected residual RNA within EV extracts to RNase A treatment to eliminate any potential interference on our results, there was no alteration detected in the expression levels of miR‑133a‑3p (Fig. 4E). Furthermore, upon disruption of EVs using Triton X‑100 followed by RNase A treatment to degrade RNA encapsulated within EVs, we found that the previously increased expression level of miR‑133a‑3p was reversed (Fig. 4F). Notably, the time-dependent and dose-dependent increase in miR‑133a‑3p expression was observed in recipient AC16 and HUVEC cells exposed to N-EVs (Fig. 4G), providing evidence for its encapsulation within EVs and subsequent transfer from donor to recipient cells.
miR-133a-3p influences the PANoptosis in endothelial cells and cardiomyocytes.
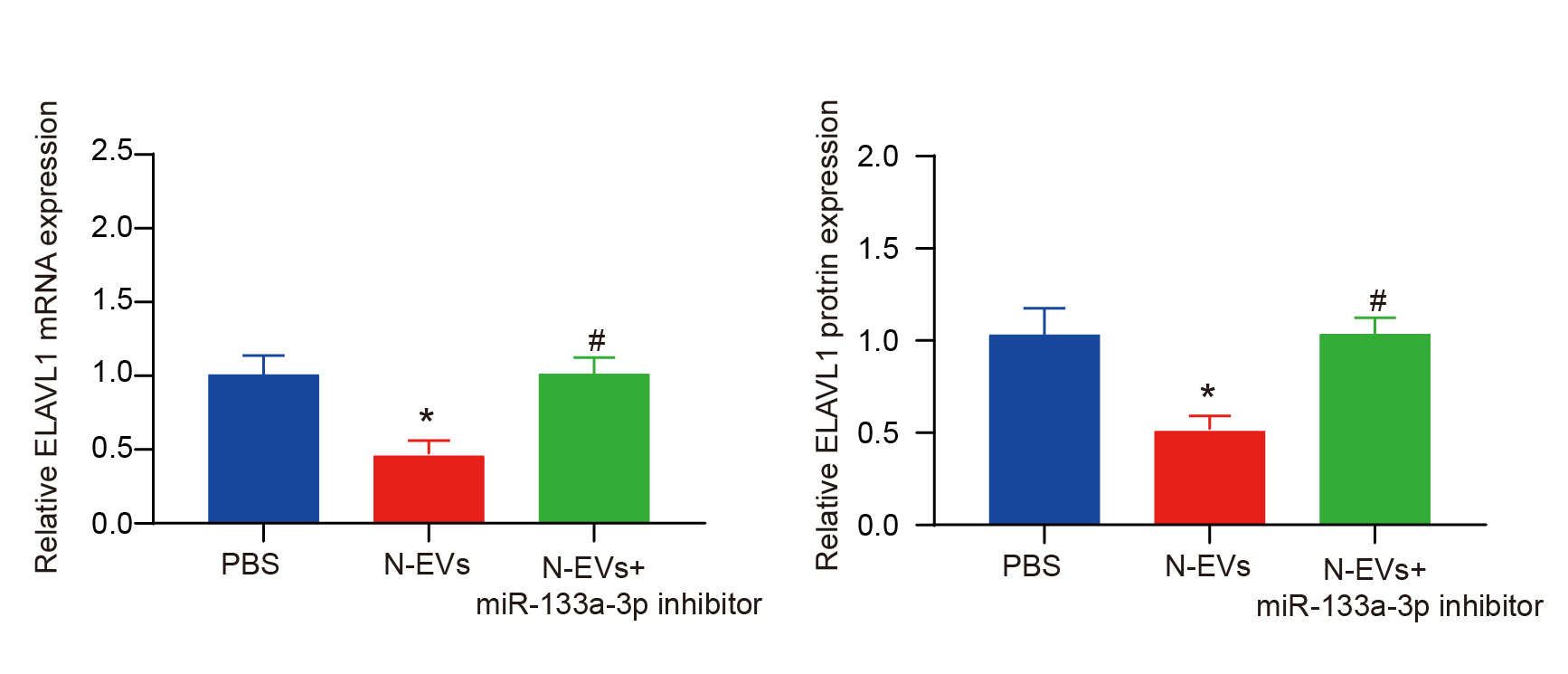
We have observed differential expression of miR-133a-3p in N-EVs. Additionally, to confirm the regulatory impact of miR-133a-3p on endothelial cells and cardiomyocytes, we conducted co-treatments using an inhibitor for miR-133a-3p and N-EVs. The outcomes from CCK8 and EdU assays revealed a significant reversal of the proliferative effect induced by N-EVs when inhibiting the expression of miR-133a-3p (Fig. 5A-B).
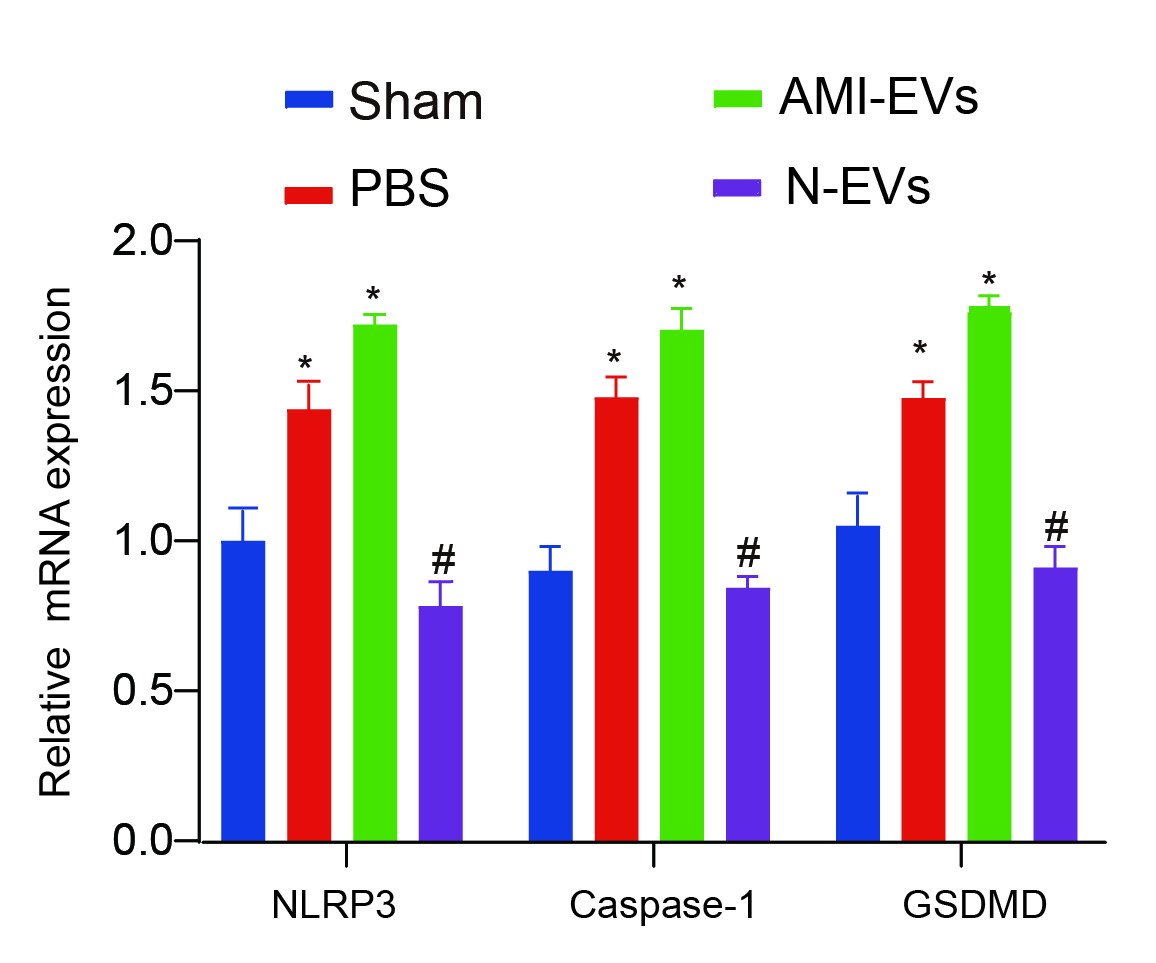
To further investigate the regulatory effect of miR-133a-3p on cell, we tested the levels of apoptosis, pyroptosis, and necroptosis in the cells. Our findings revealed a significant rise in their expression levels within EVs when miR-133a-3p was inhibited, indicating a reversal of the observed effect (Fig. 5C-F). Furthermore, our in vivo assays also confirmed that N-EVs could inhibit the level of PANoptosis in myocardial tissue (Supplementary Fig. 2).
MiR-133a-3p regulates PANoptosis by targeting ELAVL1
Currently, several studies have demonstrated the crucial involvement of ELAVL1 in PANoptosis regulation. Moreover, utilizing the starBase database, we identified a targeted regulatory relationship between miR-133a-3p and ELAVL1. Consequently, we conducted further investigations to explore their regulatory interplay. We confirmed the specific binding of miR-133a-3p to ELAVL1 through dual-luciferase reporter and RIP assays (Fig. 6A-B, Supplementary Fig. 3). Furthermore, we validated the regulatory effect of miR-133a-3p on cardiomyocyte proliferation using CCK8 and apoptosis assays, demonstrating that ELAVL1 acts as its target gene (Fig. 6C-D). Similarly, we assessed markers of PANoptosis and observed that miR-133a-3p exerts a regulatory role by modulating the expression of ELAVL1 (Fig. 6E-F).
ELAVL1 influences the formation of the PANoptosome by affecting the stability of NLRP3 mRNA.
To delve deeper into the mechanism by which ELAVL1 regulates PANoptosis, we first established ELAVL1 as an RNA-binding protein (RBP). Utilizing predictions from the starBase database, we identified ELAVL1 as an RBP specifically interacting with NLRP3 mRNA (Fig. 7A). Knocking down ELAVL1, we observed no significant changes in the mRNA expression levels of NLRP3 (Fig. 7B), yet there was a marked effect on the protein expression of NLRP3 (Fig. 7C). This led us to hypothesize that ELAVL1 influences the protein expression of NLRP3 by affecting the stability of its mRNA. Our experiments confirmed this hypothesis (Fig. 7D).
Subsequently, we employed the ATtRACT database to predict three binding sites for ELAVL1 on NLRP3 mRNA (Fig. 7E). Through luciferase reporter assays, we validated these predicted sites, pinpointing site P1 as the critical binding region through which ELAVL1 exerts control over NLRP3 (Figs. 7F-G). Additionally, RIP assays provided concrete evidence of the physical interaction between ELAVL1 protein and NLRP3 mRNA (Fig. 7H). Collectively, these findings demonstrate that ELAVL1 regulates PANoptosis by modulating the stability of NLRP3 mRNA.
{kind=link}
{kind=link}
{kind=link}