3.1 Ground State Geometry
Figure 1 shows eight phosphorescent Ir(III) complexes. The heavy-metal Ir coordinates with ligands including 2-phenylpyridine (ppy), 1-phenylisoquinolinato (piq), benzo[h]quinolinato (bzq), 2,4-pentanedione (acac), 2-(4,6-difluorophenyl) pyridinato (46dfppy), and 2-(p-tolyl) pyridinato (tpy) to form six-coordinate complexes. Four different functionals including PBE0, B3LYP, ωB97X, and CAM-B3LYP are utilized to optimize the ground-state geometries of eight molecules. Table 1 presents the structural parameters of fac-Ir(ppy)3, mer-Ir(tpy)3, and fac-Ir(piq)3 obtained from different functionals and X-ray crystallography measurements15, 29–30. Figure 1 shows the structure of the selected molecule and the key atomic labels.
Due to the C3 symmetry of the facial configuration including fac-Ir(ppy)3 and fac-Ir(piq)3, the Ir-C and Ir-N bond lengths are identical. Thus, for the facial configuration, we only analyze one Ir-C and one Ir-N bond length due to the C3 symmetry. In contrast, for the meridional configuration with C1 symmetry, we analyze all three Ir-C and Ir-N bond lengths. From Table 1, we can find that the maximum errors between calculated and experimental values in bond lengths and angles obtained from different functionals are as follows: ωB97X is 0.096 Å and 1.4 degrees, CAM-B3LYP is 0.123 Å and 1.8 degrees, PBE0 is 0.108 Å and 1.7 degrees, and B3LYP is 0.143 Å and 1.6 degrees. Compared with X-ray crystallography data15, 29–30, the computed bond length deviations are all below 0.15 Å, and the angle deviations are below 2 degrees.
These indicate that the ground state structures of the complexes obtained from DFT calculations are relatively reliable, and the choice of functional has a minor impact on the results. The minor variations in bond lengths and angles across different functionals, all remaining within acceptable error margins compared to X-ray crystallography data, underscore the robustness of DFT in modeling the ground structures of these complexes.
Given the high accuracy and consistency of the ωB97X functional, as evidenced by its minimal deviations in bond lengths and angles, it is deemed the most suitable for further studies. The ωB97X functional, with its optimal tuning and balance between short-range and long-range interactions, provides a nuanced and accurate description of the electronic environment in these complexes. Therefore, subsequent studies will employ the ground-state geometries optimized with ωB97X for further electron structure calculations, including time-dependent DFT (TD-DFT) simulations of absorption spectra.
Table 1
Comparison of the calculated bond lengths (in Å) and bond angles (in degrees) for the molecular geometry of the Ir(III) complex optimized using different functional approaches with experimental values obtained from X-ray crystallography.
| | | ωB97X | CAM-B3LYP | PBE0 | B3LYP | exp. |
| mer-Ir(tpy)3 | | Bond Lengths (Å) |
| Ir-C1 | 2.176 | 2.102 | 2.090 | 2.115 | 2.15129 |
| Ir-N1 | 2.080 | 2.167 | 2.152 | 2.187 | 2.04429 |
| Ir-C2 | 2.065 | 2.087 | 2.076 | 2.098 | 2.06529 |
| Ir-N2 | 2.103 | 2.073 | 2.057 | 2.084 | 2.07629 |
| Ir-C3 | 2.086 | 2.109 | 2.008 | 2.026 | 2.08629 |
| Ir-N3 | 2.106 | 2.058 | 2.042 | 2.066 | 2.01029 |
| fac-Ir(ppy)3 | | Bond Lengths (Å) |
| Ir-C | 2.031 | 2.033 | 2.022 | 2.041 | 2.03348 |
| Ir-N | 2.155 | 2.146 | 2.130 | 2.162 | 2.15848 |
| fac-Ir(piq)3 | | Bond Lengths (Å) |
| Ir-N1 | 2.154 | 2.145 | 2.130 | 2.161 | 2.13530 |
| Ir-C2 | 2.029 | 2.030 | 2.019 | 2.038 | 2.00930 |
| N1-C1 | 1.375 | 1.372 | 1.370 | 1.376 | 1.37430 |
| N1-C3 | 1.350 | 1.354 | 1.361 | 1.368 | 1.33930 |
| | Bond Angles (degree) |
| N1-Ir-C2 | 78.4 | 78.3 | 78.5 | 78.2 | 78.530 |
| Ir-N1-C1 | 123.4 | 123.0 | 123.1 | 123.2 | 124.830 |
| Ir-N1-C3 | 115.3 | 115.7 | 115.9 | 115.7 | 115.130 |
3.2 Effects of functionals on Frontier Molecular Orbitals (FMO)
The molecular orbital wavefunctions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of fac-Ir(ppy)3 calculated by DKH2 Hamiltonian in combination with the five different functionals including PBE0, B3LYP, ωB97X, CAM-B3LYP and optimally tuned ω*B97X are respectively showed in Fig. 2. From Fig. 2, we can see that all five functionals produce wavefunctions with the same C3 symmetric distributions for both the HOMO and LUMO. The HOMO is a mixture of d-type orbitals on the Ir atom and π-type orbitals on the three ppy ligands, while the LUMO consists of typical π-type orbitals equally distributed across the three ligands. This demonstrates that all calculated FMOs from five different DFT functionals belong to C3 symmetry.
Compared with the vertical IP values in Fig. 2, we can readily observe that the range-separated ωB97X and CAM-B3LYP functionals underestimate the energy of the HOMO, while the PBE0 and B3LYP functionals significantly overestimate it. The HOMO energy from ω*B97X is very close to the vertical IP values. In terms of the vertical EA values, the range-separated ωB97X functional overestimates the LUMO energy, while PBE0 and B3LYP notably underestimate it. The LUMO energy from the ω*B97X functional is very close to the vertical EA values. Regarding the band gap between HOMO and LUMO, ωB97X overestimates the gap, while PBE0 and B3LYP notably underestimate it. The CAM-B3LYP and ω*B97X provide similar band gaps that align well with the energy difference between IP and EA. In short summary, the tuned range-separated ω*B97X functional satisfies with Koopman's Theorem, indicating that the energies of the HOMO and LUMO are consistent with the vertical IP and EA values.
3.3 Effects of DFT functionals on 1/3MLCT
MLCT assignments
All-electron scalar relativistic DKH2 Hamiltonian combined with five DFT functionals (ωB97X, CAM-B3LYP, PBE0, B3LYP, and tuned ω*B97X) were applied to calculate the lowest excited triplet (T1) and singlet (S1) states. The electron-hole wavefunctions for the transition from S1/T1 to the ground state (S0) were analyzed using Multwfn 3.849. Using fac-Ir(ppy)3 as an example, the isosurface of electron-hole wavefunctions is shown in Fig. 3. Figure 3 demonstrates that the electron-hole wavefunctions for the S1 and T1 states exhibit DFT functional-dependent characteristics. For example, the DFT functionals B3LYP and ω*B97X produce C3 symmetric electron-hole wavefunctions, with the electron (green) and hole (blue) wavefunctions equally distributed on the three ppy ligands. The DFT functionals ωB97X and CAM-B3LYP slightly break the C3 symmetry of the electron-hole wavefunctions, while PBE0 strongly breaks the C3 symmetry, causing the electron (green) wavefunction to be primarily located on the first (I) ppy ligand.
Regardless of the electron-hole wavefunctions from different functionals for S1 or T1, the hole (blue) wavefunctions are located on the Ir atom and its surrounding ppy ligands, and the electron wavefunctions are located on the ppy ligands. The electron-hole wavefunctions for fac-Ir(ppy)3 indicate that both S1 and T1 exhibit typical MLCT electron transition characteristics. The isosurfaces of electron-hole wavefunctions for other Ir complexes, shown in Figure S1 also demonstrate similar MLCT electron transition characteristics for S1 and T1 states. Therefore, we assign the S1 and T1 states as the ¹MLCT and ³MLCT states, respectively.
Functional-dependent energies of 1/3MLCT
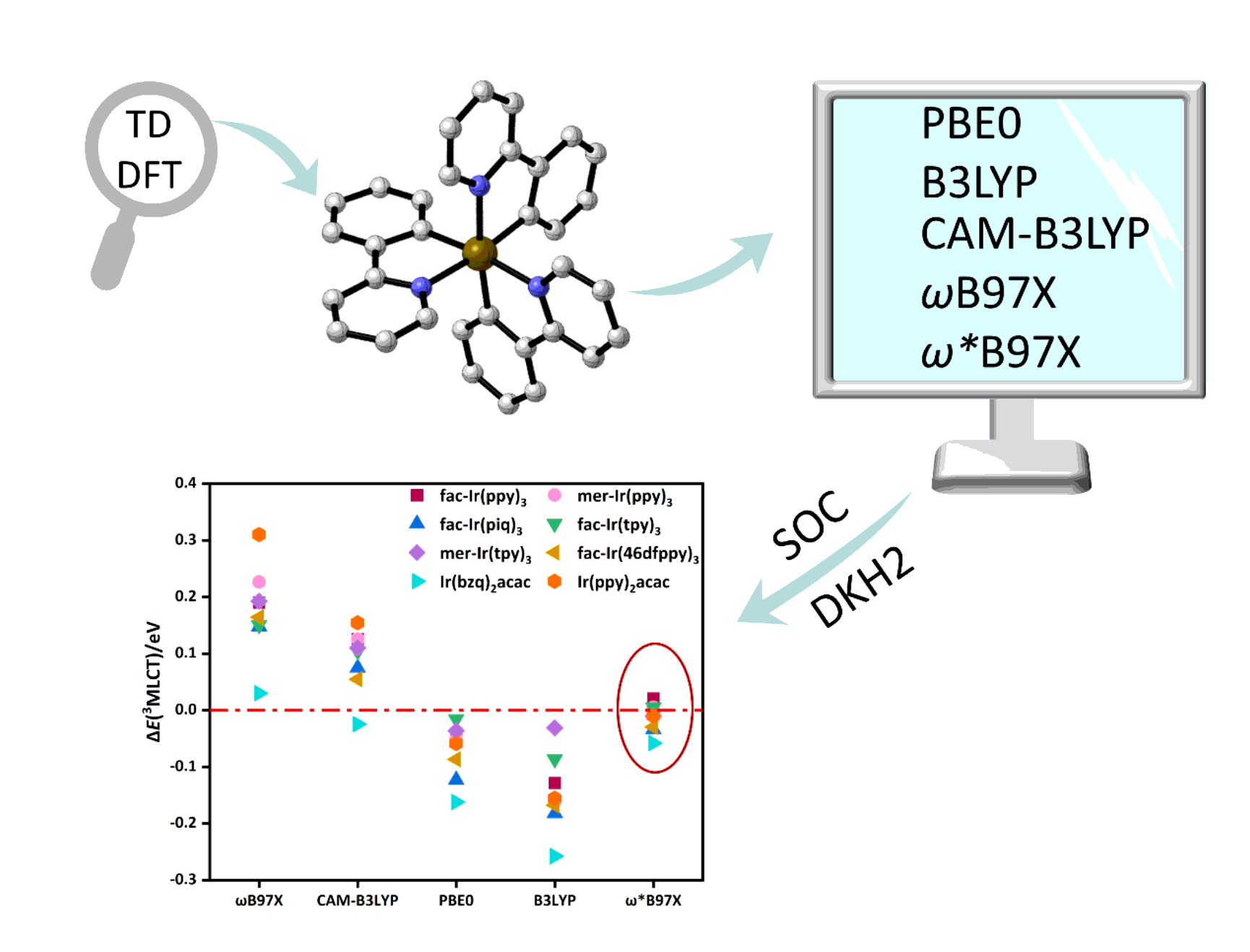
According to the energies of S1 and T1 obtained from various DFT functionals, we compared them with their respective observed 3MLCT and 1MLCT absorption peaks. The deviations (ΔE) of the calculated values from the experimental data are shown in Fig. 4 (a for ³MLCT, b for ¹MLCT). For the same DFT functional, ΔE for ³MLCT and ¹MLCT exhibits ligand-dependent characteristics.
Examining the ΔE results from the range-separated ωB97X functional, we find that for ³MLCT, Ir(ppy)2acac has the largest ΔE (~ 0.31 eV) and Ir(bzq)2acac has the smallest ΔE (~ 0.03 eV). For ¹MLCT, fac-Ir(piq)3 has the largest ΔE (~ 1.2 eV) and fac-Ir(tpy)3 has the smallest ΔE (~ 0.90 eV). Generally speaking, the range-separated functionals ωB97X and CAM-B3LYP overestimate the transition energies of 1/3MLCT, while the hybrid functionals PBE0 and B3LYP underestimate them.
The optimally tuned range-separated ω*B97X functional shows very good agreement with experimental ³MLCT and ¹MLCT, possibly because its frontier orbitals satisfy Koopman's Theorem. Looking closely at the ΔE of ¹MLCT, we can see that PBE0 also performs well in predicting ¹MLCT. Under the framework of TD-DFT, the transition energy of MLCT is mainly related to the energy gap of frontier orbitals and the two-electron correlation and exchange interaction.50 According to the analysis of frontier orbital energies, it is clear why ωB97X and CAM-B3LYP overestimate the transition energies of 1/3MLCT, while PBE0 and B3LYP underestimate them.
PBE0's good performance in predicting ¹MLCT may be due to the error cancellation effect of the overestimation of two-electron interactions and the underestimation of the energy gap. The poor two-electron interaction behavior of the PBE0 functional is evident from the previous analysis of the electron-hole wavefunction, where the electron-hole wavefunction of ¹MLCT for the C3 symmetric fac-Ir(ppy)3 notably breaks the C3 symmetry.
Considering the transition energies of ¹/³MLCT and the symmetry of the electron-hole wavefunction, we conclude that the tuned range-separated ωB97X functional performs the best in evaluating the ¹/³MLCT states of Ir(III) complexes. Therefore, for the following study of absorption spectra, we focus on the tuned range-separated ωB97X functional.
3.4 Evaluation of Higher-Energy Absorption Peaks Using the ω*B97X Functional
We have demonstrated that the ωB97X functional can accurately identify the ¹MLCT and ³MLCT energies in the absorption spectra of Ir(III) complexes. Next, we explore whether the ωB97X functional can accurately identify absorption peaks at higher energy levels in the absorption spectra, beyond the MLCT states. To assess this, we calculated the wavelengths of significant oscillator strengths using the ω*B97X functional and compared them with experimentally observed wavelengths of high absorbance peaks. Table 2 presents these comparisons and includes the mean absolute deviation (MAD) between the calculated and experimental data for each complex.
From Table 2, it is evident that the ωB97X functional not only accurately predicts the MLCT state wavelengths but also effectively models absorption peaks at higher energy levels. For instance, the maximum MAD observed is 10.7 nm for Ir(bzq)2acac, while the minimum MAD is 2.0 nm for mer-Ir(ppy)3. These small average discrepancies demonstrate the precision of the ωB97X functional in simulating high-energy absorption peaks.
The ability of the ω*B97X functional to accurately predict these higher-energy absorption peaks is significant for several reasons. First, it underscores the functional's robustness and reliability across a broader spectrum of electronic transitions, not just those limited to MLCT states. This broader applicability enhances the utility of the ω*B97X functional in computational chemistry, making it a valuable tool for predicting electronic properties in a wide range of complexes.
Additionally, accurate simulation of high-energy absorption peaks aids in the experimental synthesis of new Ir(III) phosphorescent complexes. By providing precise theoretical predictions, researchers can better design and synthesize complexes with desired photophysical properties, potentially leading to the development of more efficient and tunable phosphorescent materials.
In summary, our study shows that the ω*B97X functional not only excels in predicting ¹MLCT and ³MLCT energies but also accurately identifies higher-energy absorption peaks. This capability enhances its value as a predictive tool in computational chemistry, aiding in the design and synthesis of new Ir(III) phosphorescent complexes with optimized photophysical properties.
Table 2
Wavelengths corresponding to the large absorption intensities of the Ir(III) complexes obtained from experiments and ω*B97X functional calculations, along with the Mean Absolute Deviation (MAD) in eV between the calculated and experimental values.
| | | λabs /nm | |
| | | 3MLCT | 1MLCT | Higher MLCT | MAD |
| fac-Ir(ppy)3 | exp.29 | 488 | 455 | 405 | 377 | 341 | 283 | |
| cal. | 484 | 460 | 403 | 368 | 343 | 288 | 4.5 |
| mer-Ir(ppy)3 | exp.29 | 488 | 457 | 410 | 382 | 339 | | |
| cal. | 486 | 458 | 410 | 383 | 333 | | 2.0 |
| fac-Ir(piq)3 | exp.30 | 600 | 550 | 483 | 430 | 354 | 333 | |
| cal. | 611 | 560 | 482 | 444 | 344 | 332 | 7.8 |
| fac-Ir(tpy)3 | exp.29 | 485 | 450 | 410 | 374 | 347 | | |
| cal. | 484 | 459 | 403 | 366 | 344 | | 5.6 |
| mer-Ir(tpy)3 | exp.29 | 485 | 451 | 420 | 383 | 336 | | |
| cal. | 487 | 459 | 416 | 384 | 336 | | 3.0 |
| fac-Ir(46dfppy)3 | exp.29 | 456 | 428 | 388 | 353 | 312 | | |
| cal. | 460 | 424 | 378 | 340 | 304 | | 7.8 |
| Ir(bzq)2acac | exp.33 | 500 | 470 | 360 | | | | |
| cal. | 511 | 483 | 368 | | | | 10.7 |
| Ir(ppy)2acac | exp.33 | 497 | 460 | 412 | 345 | | | |
| cal. | 498 | 464 | 410 | 338 | | | 3.5 |
3.5 Relativistic Effects of Ir Complexes
In addition to the choice of functionals, the relativistic effects of Ir(III) complexes, including SOC and scalar relativistic effects, significantly impact the absorption spectra.
Spin-Orbit Coupling (SOC) Effects
Figure 5 shows the absorption spectra of fac-Ir(ppy)3 calculated using the ω*B97X functional coupled with DKH2 Hamiltonian. The red curve represents the absorption spectra with SOC, while the blue curve represents the spectra without SOC. As we know, SOC becomes more pronounced with increasing atomic number. For Ir(III) complexes, SOC strongly influences the splitting and mixing of electronic states. This is particularly relevant for MLCT transitions, where SOC can mix singlet and triplet states, affecting the absorption and emission spectra. Ignoring the SOC effect prevents the mixture of triplet and singlet states, making the ³MLCT transition from S0 to T1 forbidden according to spin symmetry. Therefore, we can observe that the ³MLCT absorption peak at the low energy level of the absorption spectra disappears when the SOC effect is not considered, leaving only the ¹MLCT absorption peak. The red shift ~ 0.21eV of 1MLCT for the calculation including SOC is due to strong SOC affecting on the energy levels in fac-Ir(ppy)3 molecule. The similar situations are happened on other Ir(III) complexes shown as in Figure S2.
Scalar Relativistic Effects
Relativistic effects shift the energy levels of orbitals, which can alter the electronic structure of the molecules. For Ir atom, the relativistic stabilization of the 6s orbital and the destabilization of the 5d orbitals are significant, which results in decreasing energies of inner molecular orbitals and increasing the energies of occupied valance orbitals in Ir complexes.51 The scalar relativistic effects can usually be considered by effective core potential (ECP) and all-electron relativistic approaches. To demonstrate this effect, we respectively calculate the absorption spectra of Ir complexes with all-electron DKH2 and without DKH2 Hamiltonian. As a contrast, Def2-TZVP ECP basis set also is applied to calculate the fronter molecular orbitals of Ir(III) complexes. Figure 6 shows the absorption spectra and HOMO-LUMO orbitals with and without the DKH2 Hamiltonian for fac-Ir(ppy)3, as well as the fronter orbitals using ECP method. The black curve represents spectra without the DKH2 Hamiltonian correction, and the red curve represents spectra with the DKH2 Hamiltonian correction, both accounting for SOC effects.
From the absorption spectra in Fig. 6(a), we observe that the high-energy level absorption peaks are similar with and without scalar relativistic effect, but the absorption intensities differ. However, for the lower-energy MLCT states, the spectrum without the DKH2 Hamiltonian correction is notably blue-shifted, affecting the accuracy of the absorption spectra. The similar situations are happened on other Ir(III) complexes shown as in Figure S3. Figure 6(b) shows that the energy levels of HOMO are similar with each other when the relativistic effect is considering by DKH2 Hamiltonian or ECP, while the HOMO energy without DKH2 indeed is significantly lower around 0.24eV. The smaller HOMO-LUMO energy gap for DKH2 method results in the red-shift MLCT energy transition compared with all-electron non-relativistic DFT calculation.
In summary, both SOC and scalar relativistic effects are crucial for accurately modeling the electronic structure and absorption spectra of Ir(III) complexes. Ignoring these effects can lead to significant deviations from experimental results, underscoring the importance of including relativistic corrections in computational studies of transition metal complexes.
{kind=link}