Given that the soft tissue damage and alveolar bone loss caused by periodontitis are primarily due to an excessive immune-inflammatory response triggered by microorganisms, exploring the pathogenic mechanisms of periodontitis from an immunological perspective, in addition to controlling plaque, may offer promising avenues for effective treatment32. The complement system, as a critical component of the immune system, serves as the first line of defense against pathogenic microorganisms and bridges innate and adaptive immunity33. Therefore, it is likely to play a significant role in the onset and progression of periodontitis induced by periodontal pathogens. In this study, we employ single-cell transcriptome analysis complemented by rigorous experimental validation to elucidate that the hydrolysis product of the central component of the complement system, C3a, participates in the onset and progression of periodontitis by promoting macrophage M1 polarization and osteoclast differentiation. It concludes by suggesting the potential viability of treating periodontitis by inhibiting the function of C3a or reducing local C3a levels in periodontal tissues.
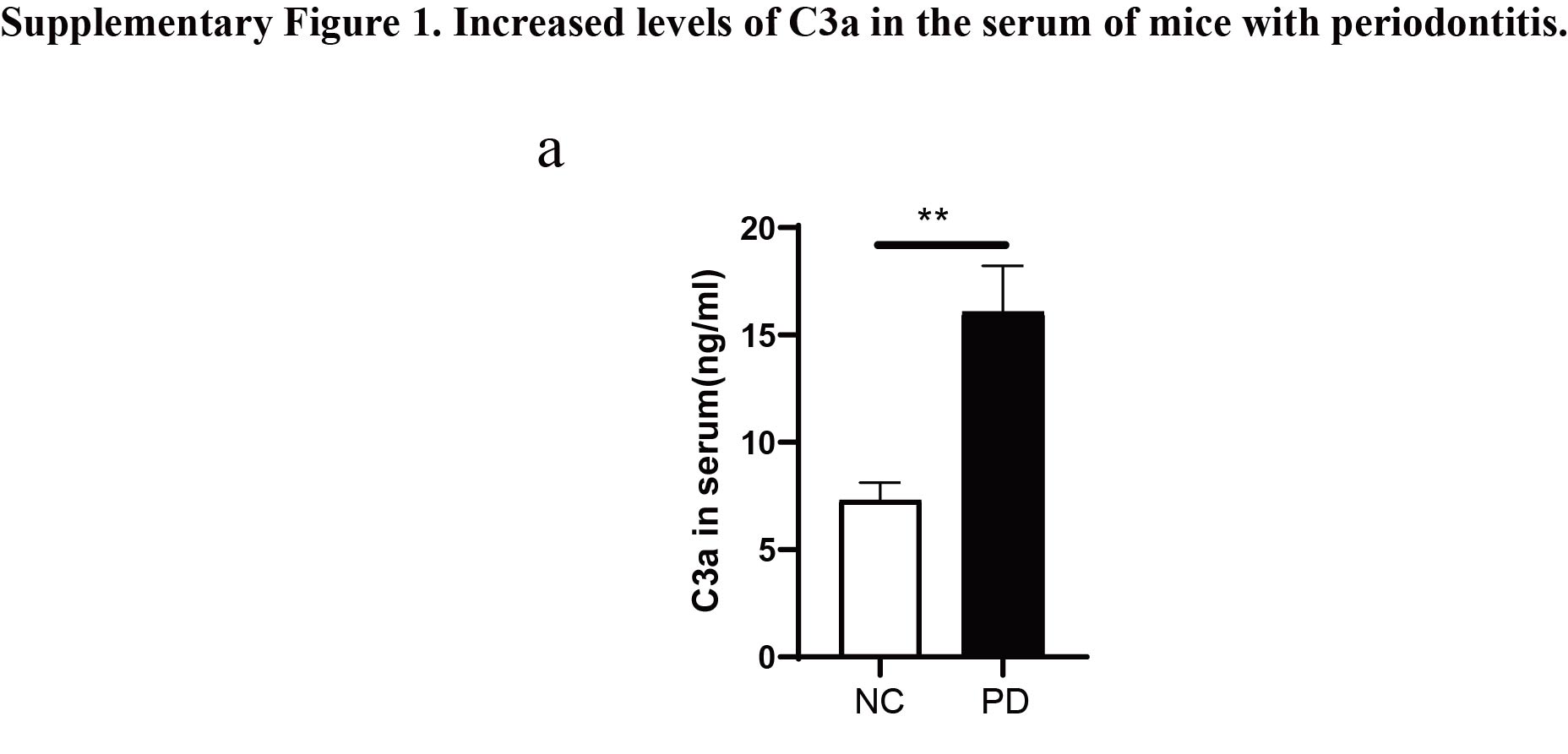
The complement system, an integral part of the immune system, has been implicated in the pathogenesis and progression of periodontitis. Early clinical studies have demonstrated significantly higher levels of complement activation products in the gingival tissue and gingival crevicular fluid of patients with periodontitis compared to healthy individuals34. The induction of experimental gingivitis in human volunteers resulted in progressive complement activation, correlating with increased clinical inflammation35. Conversely, successful periodontal treatment that resolved clinical inflammation inhibited complement activation in the gingival crevicular fluid of treated patients36. Specifically, interception of the complement cascade at its central component, C3, using a locally administered small peptidic compound (Cp40/AMY-101) protected non-human primates from induced or naturally occurring periodontitis37. Consistent with these findings, our study also found that the protein level of complement C3a in the serum of periodontitis mice was significantly higher than that in non-periodontitis mice. This suggests that complement C3a plays a crucial role in the development and progression of periodontitis.
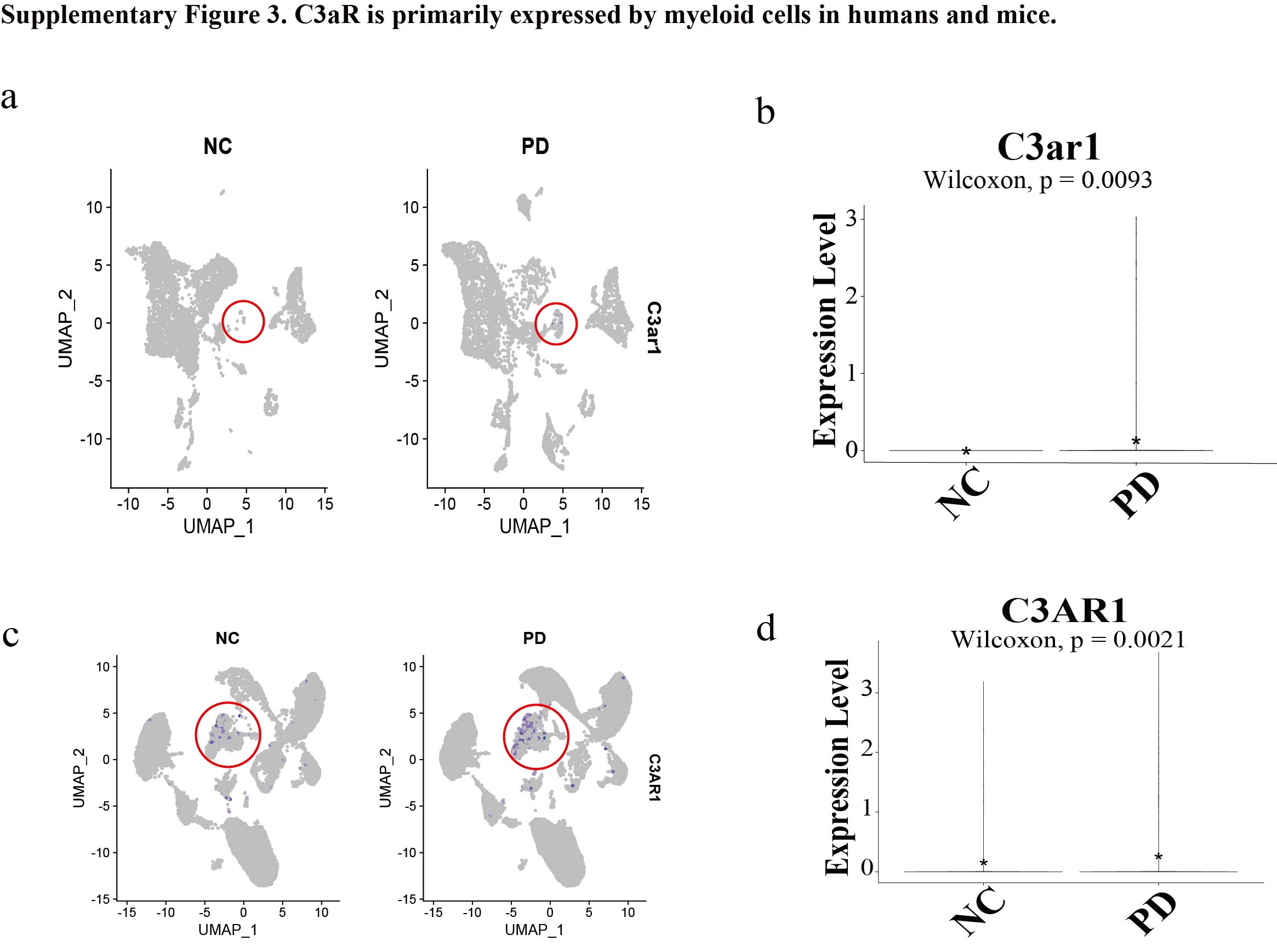
Complement has been considered mainly existing in the systemic compartment, with serum levels of most components of the complement system, including C3, C4, and MBL, being produced by hepatocytes38. Other tissues also contain cells capable of complement production; for example, endothelial and epithelial cells can secrete various complement components, thereby contributing to local processes of diseases39. Additionally, there is a growing body of evidence that local secretion of complement proteins plays an important role in regulating physiological processes even in the absence of further complement activation40. Our findings also support this view. Through single-cell sequencing data analysis of healthy and periodontitis tissues in humans and mice, we discovered that periodontal fibroblasts are the primary source of local C3 in periodontitis. Upon activation of the complement system, C3 is hydrolyzed into C3a and C3b. C3b promotes phagocytosis of bacteria by binding to corresponding receptors on phagocytic cells, or forms C5 convertase complexes with other complement components to participate in the complement cascade reaction. C3a is released and binds to its specific receptor, C3aR, on cell surfaces to exert its effects. Research indicates that numerous cell types express C3a and its receptor C3aR, including various immune cells (such as monocytes, neutrophils, macrophages, T cells, B cells, and mast cells), osteoblasts, osteoclasts, and their precursor cells41. Among these, macrophages are the primary cell type expressing C3a and C3aR within immune cells42. Indeed, in our study, we found that macrophages are the main cell type expressing C3aR. Notably, after the onset of periodontitis, the expression of the C3 gene in periodontal fibroblasts and the C3ar1 gene in macrophages both significantly increased. Furthermore, the complement system's hydrolytic pathways are enhanced, as evidenced by our single-cell data cell ratio analysis revealing a significant increase in plasma cell secretion of antibodies in the periodontitis group. This augmentation potentially strengthens the pathway involving complement activation by antigen-antibody complexes, thereby leading to increased hydrolysis of C3 into C3a. This further underscores the importance of C3a in the pathogenesis and progression of periodontitis.
The interaction between C3a and its receptor C3aR mediates a series of biological effects. For instance, this interaction regulates ATP efflux and promotes the activation of the NOD-like receptor protein 3 (NLRP3) inflammasome by increasing the phosphorylation of extracellular signal-regulated protein kinases 1/2 (ERK1/2), leading to the secretion of the inflammatory cytokine IL-1β by human monocytes, macrophages and dendritic cells43. Additionally, C3a promotes mast cell degranulation and chemokine secretion through the regulation of Ca2+ release, playing a role in hypersensitivity reactions44. This axis also facilitates skeletal muscle repair after injury by chemotactically recruiting macrophages to the site of muscle damage45. Furthermore, the C3a promotes M1 macrophage polarization through signaling pathways involving ERK1/2, nuclear factor-κB (NF-κB), and signal transducer and activator of transcription 1 (STAT1), accelerating renal fibrosis in a mouse model of unilateral ureteral obstruction19. In line with these findings, our study demonstrates that the C3a increases the number of M1 macrophages and the expression of M1 macrophage markers in periodontal inflammatory tissues. This further substantiates the role of the C3a in regulating macrophage polarization and suggests that it may mediate periodontal inflammation and soft tissue destruction by modulating macrophage polarization. These findings suggest that the heightened C3a level in periodontitis patients is more than a biomarker of disease severity, but rather reflects a critical destructive factor.
Given that a primary characteristic of periodontitis is alveolar bone loss, and osteoclasts are the only cells in periodontal tissues capable of causing this bone destruction, investigating the reasons behind the increased presence of osteoclasts in periodontal tissues is crucial. Research indicates that M1 macrophages secrete various inflammatory cytokines, such as IL-6, IL-1, TNF-α, and prostaglandin E2 (PGE2), which stimulate osteoclast differentiation and activation, thereby contributing to bone resorption46. Additionally, M1 macrophages can induce the differentiation of Th17 cells, which express high levels of RANKL, further promoting osteoclastogenesis and alveolar bone destruction47. Our findings show that knockout of C3aR decreases the number of M1 macrophages and mitigates alveolar bone loss in mice with periodontitis. This supports the hypothesis that the C3a may regulate osteoclast activity and subsequent alveolar bone destruction by modulating macrophage polarization. Furthermore, our research provides insight into another mechanism where the C3a directly promotes monocyte differentiation into osteoclasts. Recent studies in bone immunology have corroborated our findings. Anita Ignatius et al.'s research found that C3a and C5a alone were insufficient to induce the release of inflammatory cytokines IL-6 and IL-8 from osteoblasts. However, co-stimulation with the pro-inflammatory cytokine IL-1β significantly enhanced IL-6 and IL-8 expression, along with RANKL and osteoprotegerin (OPG) expression, suggesting that complement may modulate the inflammatory response of osteoblasts in a pro-inflammatory environment48. This interaction may influence osteoblast-osteoclast interactions and osteoclastogenesis, even in the absence of RANKL and M-CSF, thus indicating a direct regulatory role for complement in osteoclast formation.
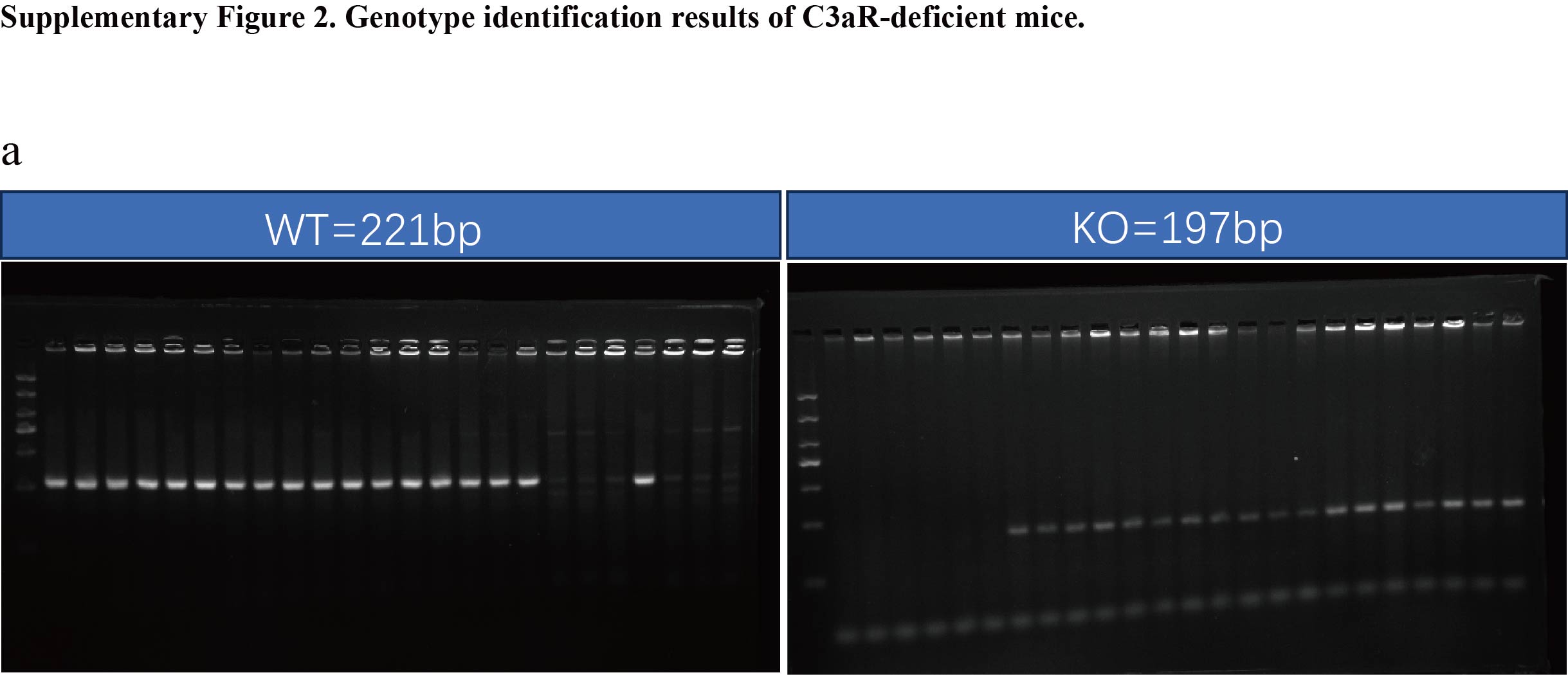
Our study partially elucidates that the C3a exacerbates periodontitis by promoting macrophage polarization and osteoclast differentiation. However, there are certain limitations to our research. Firstly, since our transgenic mice are global C3aR knockouts and we did not utilize myeloid cell-specific C3aR knockout mice, it is not entirely clear whether the amelioration of periodontitis phenotypes observed in C3aR knockout mice can be solely attributed to macrophage polarization and osteoclast differentiation. Secondly, inflammatory cytokines produced by M1-polarized macrophages can also induce osteoclast differentiation. Therefore, it remains uncertain whether the reduced alveolar bone destruction observed in C3aR knockout mice is primarily due to the decreased number of osteoclasts directly caused by the absence of C3a or the reduced inflammatory cytokine production by macrophages. Thirdly, we did not further elucidate the detailed mechanisms by which the C3a promotes M1 polarization and osteoclast differentiation.
{kind=link}
{kind=link}
{kind=link}