Data Collection and Pre-processing
Transcriptome profiling data (Count) of TGCT was downloaded from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/). Seminoma samples were included and divided into relapse (R) and non-relapse (NR) groups after reviewing the clinical data. The RNA-seq data and clinical information of the independent external validation cohort (GSE99420) was obtained from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) dataset. Single cell RNA-seq (scRNA-seq, GSE197778) data was also downloaded from the GEO dataset. Normal testis samples were obtained from the Genotype-Tissue Expression (GTEx, www.gtexportal.org/) dataset. Clinical data of chemical response was obtained from the Cbioportal dataset (MSK, J Clin Oncol 2016)8. The raw datasets, after calibration and normalization, were all converted to log2 forma using algorithm in R.
Identification of immune-related hub genes
The immune-related gene lists were obtained from ImmPort and InnateDB databases. After standardization of the expression matrix using “normalizeBetweenArrays” package in R, the differential expression immune related genes (IRGs) between seminoma and normal testis were explored with “limma” package and displayed with “pheatmap” package. |Log2Fold change (FC)| > 2 and p-value < 0.01 were set as the criteria for identifying differentially expressed IRGs.
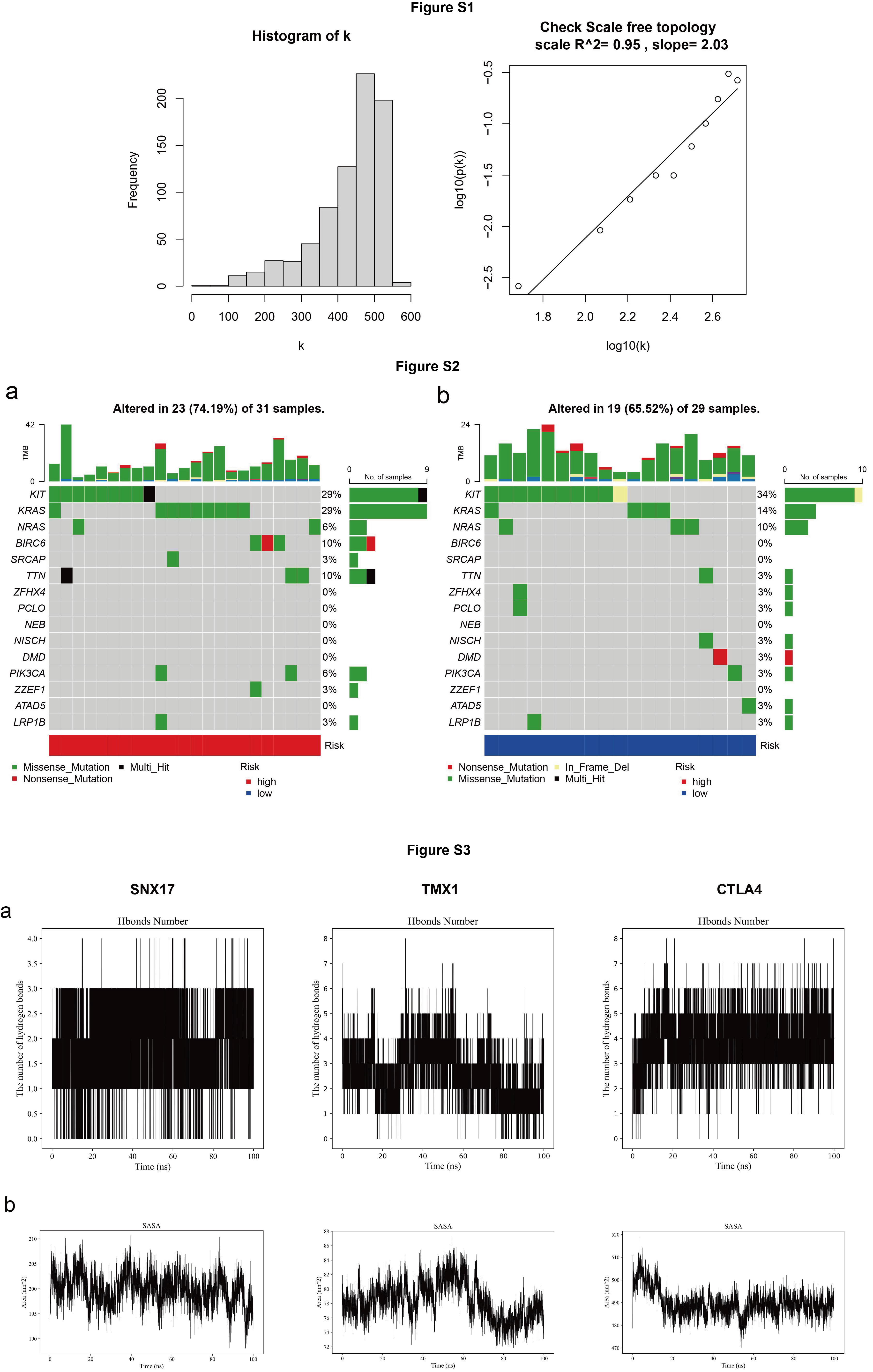
Weighted Gene Co-expression Network (WGCNA) was then performed using the “WGCNA” package in R. First, the mean expression of each gene (meanFPKM) was determined, and the genes with meanFPKM < 0.5 were removed. Second, the gene expression matrix was filtered by the “goodSamplesGenes” function to check missing values. Third, adjacency was computed using the “soft” thresholding power. The adjacency was then converted into a topological overlap matrix (TOM). The gene ratio and dissimilarity were determined subsequently. Afterward, the hierarchical clustering and dynamic tree cut functions were used to detect the modules. With a minimum gene group size (n = 50) and a TOM-based dissimilarity metric, genes with identical expression profiles were classified into gene modules using average linkage hierarchical clustering. Finally, the dissimilarity of module eigengenes was computed, a cut line for the module dendrogram was chosen, and several modules were combined for further investigation.
The String dataset (https://cn.string-db.org/) was used to visualize the network between the most significant module genes, and the Molecular Complex Detection (mcode), a plugin for Cytoscape (V3.9.0), was applied to construct the key-clusters in the network. The criteria for selection were set as follows: degree cut-off = 2, maximum depth = 100, node score cut-off = 0.2, and K-score = 2. The genes in the key cluster were considered as hub-genes.
We performed Gene set enrichment analyses (GSEA) to define the hub-genes in the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) respectively. Statistical significance was set at P < 0.05 and false discovery rate (FDR) q < 0.05. The procedure was performed using R packages including “clusterProfiler”, “org.Hs.eg.db”, “circlize”, and “dplyr”.
Immune-related Prognostic Signature (IRPS) Construction and Validation
All of the hub-genes were performed with progression free survival (PFS) analysis, and the survival related genes were deemed eligible for model construction. We conducted univariable COX proportional hazards regression analysis, lasso regression and multivariable COX regression analysis in order to construct the prognostic model. The p value filter for univariable COX regression was 0.05 and significant genes were further enrolled to conduct lasso regression. The Akaike information criterion (AIC) values were used to optimize the multivariable COX model and the RNAs with the lowest AIC were retained in the final signature. The risk score formula was as followed: Risk score= ∑Gene(exp)×Gene(coef) (Gene(exp) indicates the expression of every single RNA and the Gene(coef) was calculated using a multivariable COX proportional hazards model). Samples were defined as high-risk samples or low-risk samples based on the median risk score. The differential expression analyses of the immune-related signature genes (IRSGs) between seminoma and normal testis, R and NR were performed with “limma” package.
Kaplan-Meier curve and log-rank test were conducted to evaluate the relationship between PFS and risk score, based on TCGA-TGCT dataset and external validation cohort GSE99420 dataset.
Univariable and multivariable COX regression analyses were performed to assess the risk factors of age, stage, T, Lymphovascular Invasion (LI) and risk score in seminoma patients. The ROC analyses were subsequently performed to assess the signature’s prognostic value based on PFS years and risk factors (age, stage, T, LI, and risk score). Concordance index (C-index) was calculated with “dplyr”, “rms” and “pec” packages in R software. “Regplot”, “rms” and “survival” packages were used to establish the nomogram. “ggDCA” package was used to perform the Decision Curve Analysis (DCA). Principal components analysis (PCA) was performed based on TCGA-TGCT and GSE99429 datasets respectively using the R “scatterplot3d” package.
CIBERSORT and Immune Therapy Response
CIBERSORT was used to assess the infiltration pattern of 22 tumor immune cells in seminoma patients. The difference of immune cells infiltrating level between high and low risk groups was compared with Wilcoxon rank-sum test. For correlation test between IRSGs and the infiltration of immune cells, Spearman test was utilized. The differences between high and low risk groups in immune functions were performed with “GSVA” package. Furthermore, the tumor immune dysfunction and exclusion (TIDE) tool (http://tide.dfci.harvard.edu/) from the Harvard University was used to assess the clinical efficiency of immune checkpoint inhibition therapy. The higher TIDE predictive scores correlate with poor therapeutic effect and worse prognosis.
scRNA-seq Analysis
We performed an analysis of single-cell RNA sequencing (scRNA-seq) data using R packages, including "Seurat" and "SingleR". To ensure high-quality data, three filtering criteria were applied to each cell: (1) genes expressed in at least 5 single cells were retained, (2) cells with more than 5% mitochondrial genes were excluded, and (3) cells expressing fewer than 100 genes were removed. Initially, the scRNA-seq data underwent normalization using the "LogNormalize" method. The normalized data were then transformed into a Seurat object, and the top 1,500 highly variable genes were identified using the "FindVariableFeatures" function. Subsequently, the "RunPCA" function from the "Seurat" R package was employed to conduct principal component analysis (PCA), reducing the dimensionality of the scRNA-seq data based on the top 1,500 genes. Significant principal components were identified using JackStraw analysis, and the first 15 principal components, selected based on the proportion of variance explained, were utilized for cell clustering. Cell clustering analysis was performed using the "FindNeighbors" and "FindClusters" functions within the "Seurat" package. A k-nearest neighbor graph was constructed based on Euclidean distance in PCA, with the "FindNeighbors" function determining the closest neighbors of each cell. Subsequently, t-distributed stochastic neighbor embedding (t-SNE) was conducted using the "RunTSNE" function to visualize cell clustering, represented in t-SNE-1 and t-SNE-2 dimensions. Differential expression analysis was carried out using the "FindAllMarkers" function within the "Seurat" package, employing Wilcoxon–Mann–Whitney tests to identify differentially expressed genes (DEGs) for each cluster. Marker genes for each cluster were determined based on cutoff threshold values, with an adjusted p-value < 0.01 and |log2 (fold change) | >1. For cluster annotation, a reference-based annotation approach was employed, utilizing reference data from the Human Primary Cell Atlas.
Sample collection
Seminoma samples and adjacent normal tissues were obtained from 38 patients (mean age: 34 ± 4 years, range: 21–43 years) between 2011 and 2021 at the First Affiliated Hospital of Nanjing Medical University and Nanjing Jinling Hospital. These patients underwent orchiectomy and were subsequently diagnosed with testicular seminoma based on postoperative pathology. Among them 12 patients suffered relapse after surgery (7 of them locoregional recurrence; 4 of them new primary tumor, 1 of them distant metastasis). Samples for RNA extraction were promptly frozen in liquid nitrogen and stored at -80°C. Samples intended for immunohistochemical analysis were fixed in formalin. The study design and protocol were ethically approved by the ethics committee of the Jinling Hospital, and informed consent was obtained from all participants.
RNA extraction, reverse transcription, and quantitative RT-PCR
Total RNA was extracted from testis tissues and cell lines using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The total RNA was reverse-transcribed into complementary DNA (cDNA) using HiScript II (Vazyme, Shanghai, China). qRT-PCR was performed using SYBR Green I (Vazyme, Shanghai, China) on ABI 7900 system (Applied Biosystems, Carlsbad, CA, USA) and the primers were as followed: CTLA4 Forward: 5’-GCCCTGCACTCTCCTGTTTTT-3’; CTLA4 Reverse: 5’-GGTTGCCGCACAGACTTCA-3’. SNX17 Forward: 5’- CGCCTACGTGGCCTATAACAT-3’; SNX17 Reverse: 5’- CAATGGGTCTTGCCGAACAG-3’. TMX1 Forward: 5’- TTGCGAAAGTAGATGTCACAGAG-3’; TMX1 Reverse: 5’- CTGATAGCGCCTAAATTCACCAT-3’.
Immunohistochemistry (IHC) and immunofluorescence (IF)
IHC staining and evaluation followed previously established protocols19. Specifically, protein expression levels of CTLA4, SNX17, and TMX1 in both seminoma and adjacent normal testis tissues were assessed using anti-CTLA4, anti-SNX17, and anti-TMX1 antibodies, respectively.
For tissues representing both relapse and non-relapse seminomas, coverslips were initially washed three times in PBS, followed by fixation in 4% paraformaldehyde for 15 minutes. Subsequently, permeabilization was achieved by treating slides with 0.5% Triton X-100 at room temperature for 20 minutes, followed by three PBS washes and blocking with 2% BSA for 30 minutes. Slides were then incubated overnight at 4°C with the appropriate primary antibodies, followed by incubation with secondary antibodies at room temperature for 2 hours. Nuclei were counterstained with DAPI, and the slides were scanned using a 3DHISTECH scanner (Pannoramic MIDI).
Cell culture
The Tcam-2 cell line, derived from testicular seminoma, was procured from the Department of Urology, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine. The cells were cultured in DMEM medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY) at 37°C in a 5% CO2 atmosphere.
Antibodies and reagents
Primary antibodies were obtained from: Mouse anti-CTLA4 (Santa Cruz, sc-376016), Rabbit anti-SNX17 (abcam, ab223046), Rabbit anti-TMX1 (abcam, ab244263), anti-GAPDH, CD27, CD22, CD3, Vimentin, E-cad, N-cad, Ki-67 were all obtained from Servicebio technology. Secondary antibodies including anti-mouse IgG, anti-rabbit IgG, Alexa Fluor Secondary antibody (Spectrum 440- aqua, 488-green, 546-red, 594-orange, 647-yellow) were also gained from Servicebio technology. Rutin, ICG-001, and Doxorubicin were obtained from Selleck chem.
Cell proliferation assay and colony formation assay
To assess cellular proliferation, Tcam-2 cells were seeded at a density of 2 × 10^3 cells per well in 96-well plates. The CCK-8 system (Dojindo, Japan) was subsequently added to each well at 0, 1, 2, 3, and 4 days of culture following the manufacturer’s instructions. Absorbance readings at 450 nm were taken using a microplate reader (Tecan, Switzerland) to determine cell viability.
For the colony formation assay, approximately 1 × 10^3 cells were plated in six-well dishes and treated with DMSO, Rutin, ICG-001, and Doxorubicin, respectively. After two weeks of incubation, colonies were fixed with paraformaldehyde, stained with 0.1% crystal violet, and subsequently imaged and counted.
Wound healing and transwell assay
Cells were seeded into six-well plates at a density of 3 × 10^5 cells per well and cultured until reaching 90–100% confluence. Following scratching with a 10-uL pipette tip, wells were washed twice with PBS to eliminate cell debris and residual serum. Subsequently, cells were cultured in serum-free DMEM for an additional 12 hours. The wound edges were photographed using DP-BSW software (10× objective) 12 hours post-injury and measured using ImageJ software.
For transwell assays, cells were seeded at a density of 1 × 10^5 cells in 200 µL of serum-free medium onto the upper chamber with an 8-mm pore size, either coated or uncoated with 50 µL of Matrigel (BD Biosciences). The lower chambers were filled with DMEM containing 20% FBS as a chemoattractant. Following incubation at 37°C for 12 hours for migration and 48 hours for invasion, cells that had migrated to the underside of the membrane were fixed in 4% paraformaldehyde for 30 minutes and subsequently stained with 2% crystal violet. Non-migrating or non-invading cells were gently removed, and cells that had migrated to the outer chamber were counted in five representative fields at 200× magnification.
Western blotting
Cells treated with reagents were lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China). The harvested proteins were quantified using the bicinchoninic acid (BCA) kit (Beyotime Biotechnology), separated on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto polyvinylidene fluoride (PVDF) membranes (Sigma-Aldrich, St. Louis, MO). These membranes were then blocked in Tris-buffered saline (TBS) containing 5% non-fat milk for 2 hours. Following overnight incubation with primary antibodies at 4°C, the membranes underwent three washes with TBS containing 0.1% Tween 20 (TBST). Subsequently, they were incubated with secondary antibody solution at room temperature for 2 hours. Following another round of washing, the signals were detected using a chemiluminescence system and analyzed with Image Lab software.
Xenografts in mice
Tcam-2 cells were harvested and mixed with Matrigel in a 1:1 ratio. The cell mixture was then injected into the testes of male nude mice aged 4–6 weeks, with five mice per group and a total of four groups. DMSO, Rutin, ICG-001, and Doxorubicin were administered intravenously via the tail vein once a week. After five weeks, the mice were euthanized, and the tumor masses were promptly excised for immediate measurement of weight and volume. Tumor dimensions (length L and width W) were recorded to calculate tumor volume (V) using the formula: V = (L * W^2) / 2.
Molecular compound screening and docking
The protein crystal structures of the three IRSGs were downloaded from the PDB dataset. The candidate molecular structures were downloaded from the Pubchem dataset (Table S1). The Molecular Operating Environment (MOE) software (version 2019.0102) was utilized to prepare protein structures and conduct docking scoring. In brief, the active site was determined for each enzyme using the MOE alpha site finder. Ligand molecules were then positioned within the site using the Triangle Matcher method and ranked utilizing the London dG scoring function. Subsequently, the ten best poses (with a default of 30) were selected and subjected to further refinement through energy minimization within the binding pocket. Following refinement, rescoring was performed using the GBVI/WSA dG scoring function.
Molecular dynamics simulation
The molecular dynamics simulations utilized Gromacs 2022.3 software13. Initial preprocessing of small molecules involved the application of AmberTools22 to apply the GAFF force field and Gaussian 16W for hydrogenation and calculation of RESP potential. Subsequently, the resulting potential data were integrated into the topology file of the molecular dynamics system. Simulations were conducted under standard conditions, maintaining a static temperature of 300 K and atmospheric pressure (1 bar). The Amber99sb-ildn force field was employed, with water molecules serving as the solvent (Tip3p water model), and the overall charge of the simulation system was neutralized by the addition of an appropriate quantity of Na + ions. The energy minimization stage utilized the steepest descent method, followed by equilibration in the isothermal isovolumetric ensemble (NVT) and isothermal isobaric ensemble (NPT), each for 100,000 steps, with a coupling constant of 0.1 ps and a duration of 100 ps. Subsequently, free molecular dynamics simulations were conducted, spanning 5,000,000 steps with a step length of 2 fs, resulting in a total duration of 100 ns. Post-simulation analysis involved the utilization of the built-in tools within the software to compute various parameters including root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and the protein rotation radius for each amino acid trajectory. Additionally, free energy calculations, such as MMGBSA, and free energy topography were performed, integrating the trajectory data with other pertinent analyses.
Statistical analysis
The majority of analyses were conducted utilizing R software (version 4.0.0) or Perl scripts (version 5.32.0). Perl scripts were primarily employed for data processing tasks, including gene ID transformation and data merging. Group comparisons were conducted using the Student’s t-test or chi-square test, as appropriate for the comparison of two or three groups. A significance threshold of p < 0.05 was applied, unless specified otherwise.
{kind=link}
{kind=link}