Cell lines and mice
Human SHED cells (P2) were obtained from Saliai Stem Cell Science and Technology Co., Ltd. (Guangzhou, China). Human ADSC cells (P2) and human USC cells (P2) were obtained from Cyagen Biosciences (Guangzhou, China). IPSCs were obtained from MEGAROBO Technology Co., Ltd. (Beijing, China). CaCO2 cells were preserved by cryopreservation in our laboratory. MSCs were cultured in OptiVitro® MSC Expansion Medium XF (ExCell Bio, Suzhou, China), IPSCs were cultured in PSCeasy hESC/hiPS Medium (CELLAPY, Beijing, China), and CaCO2 cells were cultured in DMEM (XP Biomed, Shanghai, China). All cells were routinely maintained at 37°C in humidified air containing 5% CO2. The culture medium was replaced every 3 days; when the confluence of the cells reached 80%-90%, the cells were washed with PBS (Coolaber, Beijing, China) and passaged through 0.25% trypsin-EDTA (Biosharp, Hefei, China) digestion.
Male BALB/c mice (12 months old) were obtained from SiPeiFu Biotech (Beijing, China). The mice were kept in the absence of specific pathogens, had a 12-hour light/dark cycle and were raised to 20 months of age. Mice were randomly divided into two groups, and the weights of the mice were evenly distributed among the groups. Then, the mice received an intraperitoneal injection of 30 mg/kg/day Scu or 0.9% saline solution daily for 80 days. All care and treatment of the experimental animals were in strict accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care approved by the Institutional Animal Care and Use Committee of the Chinese Materia Medica China Academy of Chinese Medical Sciences (license no. ERCCACMS21-2203-02).
Compound preparation, cell viability assay, and cell colony formation
AKG (Sigma, Louis, USA) and DM-aKG (Sigma, Louis, USA) were dissolved in pure water as stock solutions at 100 mM and stored at -20°C. Scu (Nature Standard, Shanghai, China) was dissolved in dimethyl sulfoxide (DMSO) (Sigma, Louis, USA) as a 100 mM stock solution and stored at -20°C. aKG, DM-aKG, and Scu stock solutions were freshly diluted with medium to their final concentrations before each in vitro experiment. The final DMSO concentration did not exceed 0.1%.
Cells were plated in 96-well plates and treated with the indicated compounds. After 48 hours of incubation, the cells were incubated with CCK-8 reagent (MedChemExpress, Shanghai, China) for another 2 hours. Then, the absorbance at 450 nm was determined spectrophotometrically on a Synergy2 multimode microplate reader (BioTek, Winooski, USA). Cell viability is expressed as a percentage relative to that of the control group after subtraction of the background signal. For the cell colony formation assay, MSCs were seeded in 6-well plates, treated with the indicated compounds, and stained with crystal violet solution.
SA-β-gal staining
SA-β-gal staining was used to detect cell senescence. According to the instructions of the β-gal staining kit (Solarbio, Beijing, China), the cells were washed with PBS twice, fixed for 15 min, and incubated overnight at 37°C. Images were taken randomly, and the SA-β-gal-positive cells were counted.
Detection of aKG
For the cell samples, the cells were resuspended in PBS, ultrasonically disrupted on ice, and then centrifuged at 8000 × g for 10 min at 4°C. For tissue samples, fresh tissue was homogenized in PBS on ice and then centrifuged at 8000 × g for 10 min at 4°C. All the supernatant was removed and placed on ice for testing. Serum samples can be used directly for testing. The aKG concentration was assessed using a human α-ketoglutaric acid (aKG) ELISA kit (Bohu, Shanghai, China) according to the manufacturer's instructions.
Western blotting
Whole-cell lysates were prepared using RIPA lysis buffer (Epizyme, Shanghai, China) supplemented with complete protease inhibitor cocktail (Beyotime, Shanghai, China), homogenized and centrifuged at 12000 × g for 10 min at 4°C. The protein concentration of the cell lysates was determined by BCA protein assay reagent (Solarbio, Beijing, China). The cell lysates were incubated in SDS‒PAGE sample loading buffer at 95°C for 10 min, separated by 8%-12% SDS‒PAGE, and transferred to PVDF membranes (Millipore, Billerica, USA). The membranes were blocked with 5% skim milk at 25°C for 30 min and then incubated with primary antibodies against IDH1 (1:3000, #12332-1-AP, Proteintech), IDH2 (1:1000, #A7190, ABclonal), IDH3A (1:1000, #A14650, ABclonal), IDH3B (1:1000, #A13742, ABclonal), GLS (1:1000, #A11043, ABclonal), Glud1 (1:1000, #A7631, ABclonal), Glud2 (1:1000, #A6604, ABclonal), p16 (1:1000, #A11651, ABclonal), p21 (1:1000, #A1483, ABclonal), HIF1a (1:1000, #A6265, ABclonal), Hydroxy-HIF-1α (1:1000, #D43B5, Cell Signaling Technology), RPS23 (1:1000, #BF8511, AFfirm), OGFOD1 (1:1000, #A16543, ABclonal), hydroxyproline (1:1000, #bs-10389R, Bioss), GAPDH (1:20000, #A19056, ABclonal), or β-tubulin (1:1000, #AC105, ABclonal) overnight at 4°C. Subsequently, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG secondary antibodies (Beyotime, Shanghai, China) for 1 hour at room temperature. SuperKine™ hypersensitive ECL luminescent solution (Abbkine, Wuhan, China) was used for the detection of the proteins of interest. The membranes were analyzed by an SH-520 Gel Imaging Analysis System (Shenhua Bio, Shanghai, China) and quantified by ImageJ software.
Immunofluorescence (IF) Assay
The cells were seeded onto glass coverslips (Biosharp, Hefei, China), treated with the indicated compounds for 48 hours, and fixed in 4% paraformaldehyde for 5 min. After being washed with PBS three times, the cells were permeabilized with 0.2% Triton X-100 for 3 min, blocked with 5% BSA for 60 min at room temperature, and probed with primary antibodies against Ki67 (1:300, #A21861, ABclonal), γH2AX (1:300, #AP0099, ABclonal), RPS23 (1:200, #BF8511, AFfirm), and OGFOD1 (1:200, #A16543, ABclonal) for 60 min at room temperature. Then, the cells were exposed to Alexa Fluor 594-labeled (red) anti-rabbit or FITC-labeled (green) anti-rabbit secondary antibodies (Bioss, Beijing, China) and stained with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime, Shanghai, China). Image acquisition was achieved using an EVOS M7000 intelligent imaging system (Thermo Fisher Scientific, Lafayette, USA) and a STEDYCON (Abberior, Gottingen, Germany) attached to a Olympus BX53 (Olympus, Tokyo, Japan).
TCA-targeted metabolic flux analysis
After the cells completely adhered to the wall, the old medium was discarded, and the adherent cells were gently washed twice with PBS. Then, the PBS was discarded, and the prepared D-glucose-13C6 (Macklin, Shanghai, China)-labeled medium was added to the Petri dish to continue culture. The cells were collected after 24 hours of culture. Metabolites were extracted from cells using acetonitrile: water (1:1) and derived with 3-nitrophenylhydrazine. The metabolites were analyzed by a Jasper HPLC-Sciex 4500 MD. The chromatographic conditions were as follows: Phenomenex Kinetex C18 chromatographic column (100 × 2.1 mm, 2.6 µm). Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile.
TPP
After centrifugation at 12000 × g for 10 min at 4°C, low-generation and high-generation ADSC cells were mixed at a 1:1 ratio and then lysed in PBS supplemented with 1% EDTA-free cocktail to obtain soluble proteins. The supernatant was divided into three equal parts, two of which were treated with 5 µM aKG and 40 µM aKG (dissolved in pure water), and the other was treated separately with the same amount of pure water as the vehicle. The protein extract was incubated for 30 min with aKG or pure water at room temperature, heated for 4 min at 52 ℃, and then cooled for 3 min at room temperature. The heated pyrolysis products were centrifuged at 20000 × g for 20 min at 4°C to separate soluble proteins from precipitated proteins. The collected supernatant was divided into two parts: one was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‒PAGE) to quantify the protein band strength, and the other was analyzed by TMT-based quantitative proteomics.
CETSA
For the validation of RPS23 based on western blotting analysis by TPP, two portions of ADSC cell lysates incubated with DMSO or 100 µM aKG were divided into eight aliquots. All aliquots were heated individually at different temperatures for 4 min. After cooling on ice for 3 min, the soluble fraction was obtained by centrifugation as described above and used for western blotting.
SIP
A solvent-induced protein precipitation assay was conducted as described previously 19. The two portions of ADSC cell lysate incubated with DMSO or 100 µM aKG were divided into seven aliquots. Denaturation is initiated by the addition of an acetone/ethanol/acetic acid (A.E.A.) mixture of organic solvents. When the ratio of organic solvent was 50:50:0.1, the final organic solvent ratio ranged from 9–16%. After equilibrating at 37°C for 20 min, the soluble fraction was obtained by centrifugation as described above and used for western blotting.
Quantitative RT‒PCR
Total RNA was isolated using a total RNA extraction kit (Bioss, Beijing, China). RNA was reverse-transcribed to complementary DNA (cDNA) with ABScript III Reverse Transcriptase (ABclonal, Wuhan, China). Quantitative real-time reverse transcription polymerase chain reaction (RT‒PCR) was performed using Universal Blue qPCR SYBR Green Master Mix (Yeasen, Shanghai, China). The 20 µL reaction mixture contained 400 nM primers, 10 µL of qPCR SYBR Green Master Mix, 2 µL of template cDNA, and nuclease-free water. cDNA amplification was conducted via QuantStudio™ 7 Flex quantitative real-time PCR (Applied Biosystems, Foster City, USA) following the manufacturer’s instructions. GAPDH was used as an internal control.
IDH1 activity assay
To detect the direct effect of Scu on the IDH1 protein in MSCs, IDH1 antibody plates were generated by coating IDH1 antibody (Proteintech, Wuhan, China) onto 96-well plates (100 ng/well) overnight at 4°C. ADSC lysates were added to IDH1 antibody plates, incubated at room temperature for 2 hours, and then washed with PBS to remove unbound protein to obtain IDH1 protein plates. After incubation for 1 h with varying concentrations of Scu in IDH1 protein plates, three washes with PBS were performed, and the relative IDH1 activity was detected with a Cytoplasmic Isocitrate Dehydrogenase (ICDHc) Activity Detection Kit (Solarbio, Beijing, China) according to the manufacturer's instructions.
Differential scanning fluorimetry (DSF)
A total of 10 µL of RPS23 recombinant protein (0.2 µg) was mixed with 2 µL of aKG (100 µM) or 2 µL of PBS in a reaction mixture containing 5 µL of protein thermal shift buffer and 2.5 µL of protein thermal shift dye (Thermo, Foster City, USA). The prepared reaction mixture was transferred to a 0.1 mL multistrip PCR tube. DSF was performed using QuantStudio™ 7 Flex quantitative real-time PCR (Applied Biosystems, Foster City, USA), and each melting curve was programmed as follows: 25°C for 2 min, followed by a 1°C increase per min from 25°C to 95°C and finally 95°C for 2 min.
Generation and characterization of MSCs with Gene Knockdown and Overexpression
The three highest-scoring shRNA sequences targeting human IDH1 were designed and synthesized by using the pLV-U6-SHRNA-CMV-EGFP(T2A)-PURO vector (Scilia, Beijing, China). The human IDH1 gene expression lentiviral vector was designed and synthesized by using the pLV[Exp]-mCherry:T2A:Bsd-EF1A > FLAG/hIDH1 vector (VectorBuilder, Guangzhou, China). The shRNA sequences targeting human OGFOD1 were designed and synthesized by using the pRP[shRNA]-Bsd-U6 > hOGFOD1 vector (VectorBuilder, Guangzhou, China). The IPTG-inducible shRNA sequences targeting human OGFOD1 were designed and synthesized by using the pLV[shRNA]-LacI:T2A:Bsd-U6/2xLacO > hOGFOD1 vector (VectorBuilder, Guangzhou, China). Empty vectors were used as negative controls. A Lentiviral Packaging Kit (Biorigin, Beijing, China) was used for lentiviral packaging according to the manufacturer’s protocols. The cells were then infected with lentivirus concentrate for 24 hours and cultured in MSCs expansion medium. After 72 hours, puromycin or blasticidin was used to select gene knockdown and overexpression cell lines, respectively.
Flow cytometry
CD73 (1:50, #bs-4834R-APC, Bioss), CD90 (1:100, #16897-MM10-P, SinoBiological) and CD105 (1:100, #10149-MM13-PE, SinoBiological) as positive markers and CD34 (1:100, #68035-XM01-F, SinoBiological), CD45 (1:50, #10086-MM05-F, SinoBiological) and CD116 (1:100, #A23355, ABclonal) as negative markers, which were used to characterize MSCs by a Beckman Coulter CytoFLEX (Beckman Coulter, California, USA).
Osteogenic and adipogenic
Following the manufacturer's instructions, a human-related stem cell osteogenic differentiation kit (Cyagen, Suzhou, China) and a human-related stem cell adipogenic differentiation kit (Cyagen, Suzhou, China) were used to induce the osteogenic and adipogenic differentiation of MSCs, respectively. After 25 days of osteogenesis and lipogenesis, calcium nodules and lipid droplets were observed and photographed under a microscope after staining with Alizarin Red S (Cyagen, Suzhou, China) and Oil Red O (Cyagen, Suzhou, China).
BLI analysis
The binding affinities of the compounds for recombinant RPS23 were determined by a biolayer interferometry assay using Gator Plus (Gator Bio, California, USA). The recombinant RPS23 protein was labeled with a 2-fold molar amount of biotin reagent, and unbound biotin was removed by ultrafiltration. The SMAP biosensor probe (Gator Bio, California, USA) was prewetted with kinetic buffer (PBS, 0.05% BSA, 0.01% Tween 20), and then biotin-labeled recombinant protein was loaded onto the equilibrated SMAP biosensor probe. A group of probes incubated in protein-free buffer was used as a control. All the data were analyzed by GatorBio data analysis software, and the equilibrium dissociation constant (Kd) was calculated according to the ratio of Koff to Kon.
Immunoprecipitation
Whole-cell lysates were prepared using RIPA lysis buffer (Epizyme, Shanghai, China) supplemented with complete protease inhibitor cocktail (Selleck, Houston, USA) and incubated with 40 µL of Protein G Magnetic Beads (Yeasen, Shanghai, China) bound to the corresponding antibodies at 4°C overnight. After washing with PBS three times, the immunoprecipitate was analyzed by Western blotting.
Translation fidelity dual luciferase assays for use in MSCs
To measure translation fidelity in MSCs, we used a previously published double luciferase p2luci plasmid 20. Insert GCAGGAACACAATAGCAATTACAGA as an STOP reference at the polylinker for the insertion window position between Renilla Luc and firefly Luc and insert GCAGGAACACAACAGCAATTACAGA as a no-STOP reference at the polylinker for the insertion window position between Renilla Luc and firefly Luc. These translation fidelity reports were cloned and inserted into the modified pLV-EF1A vector (VectorBuilder, Guangzhou, China). The percentage of stop codon readthrough was calculated by dividing the Firefly/Renilla ratio of the stop codon readthrough or miscombination report by the average Firefly/Renilla of the control report, as in previously published literature 16.
Protein expression and purification
RPS23 (residues 1 to 143) was cloned and inserted into the NdeI/Xho I sites of the pET-28a + vector. The recombinant plasmids were transformed into E. coli BL21 (DE3) cells (Vazyme, Nanjing, China), cultured in 300 mL of Luria–Bertani (LB) medium at 37°C until the absorbance at OD600 reached 0.4–0.6, after which the cells were induced with 0.4 mmol/L isopropyl-D-thiogalactopyranoside (IPTG) for 6 h at 16°C. To obtain the nondenatured protein, cell debris was removed by centrifugation, and the supernatant was loaded onto preequilibrated Ni-NTA resin (Beyotime, Shanghai, China). Proteins were eluted using 200 mM imidazole. Then, the imidazole solution was replaced with a PBS solution by ultrafiltration. Finally, the protein concentration was measured using a BCA kit (Solarbio, Beijing, China), and the final protein was concentrated to 10 mg/ml and stored at 4°C.
Microfluidic device fabrication
Microfluidic devices were designed and calibrated using polydimethylsiloxane (PDMS) materials (Wenhao, Suzhou, China) to construct liver organoids and intestinal barriers according to previous studies. The intestinal barrier was formed via in situ culture, and liver organoids were added after the induction of maturation. CaCO2 cells were seeded in transwell plates (Corning, NY, USA) and cultured continuously for 14 days to form the intestinal barrier. IPSC-derived liver organoid induction methods were described in previous articles 21. Quality control of the intestinal barrier and liver organoids was performed. Crystal violet (0.1%) and 100 µM 2-NBDG probe staining were used to test the integrity of the barrier. The expression of the markers CYP3A4 (1:100, 67110-1-Ig, Proteintech), ALB (1:100, 16475-1-AP, Proteintech), AFP (1:100, 14550-1-AP, Proteintech) and FOXA2 (1:100, 22474-1-AP, Proteintech) in liver organoids was tested using immunofluorescence. Liquid chromatography was used to detect the concentration differences of Scu under different metabolic modes in the culture media. After 24 hours of cyclic cultivation in the entire device, the levels of aging biomarkers of mesenchymal stem cells were detected by immunofluorescence.
JC-1 assay
All cells were stained using a JC-1 staining kit (Beyotime) following the manufacturer’s instructions. After staining, the cells were immediately examined using an EVOS M7000 intelligent imaging system (Thermo Fisher Scientific).
Micro-CT analysis
Mouse femurs were removed and placed in PBS at room temperature for testing. The distal femurs were scanned with a Micro-CT image system (SkyScan 1276, Bruker, Germany). A voltage of 60 kV, current of 200 µA, scanning resolution of 6.5 µm and visual field size of 2016*1344 were used to scan the femurs of the mice. Using the lowest end of the growth plate of the knee joint of the femur as the baseline, the bone marrow cavity region with a thickness of 1 mm was selected as the region of interest for three-dimensional reconstruction (ROI). The three-dimensional image was reconstructed with NRecon software and analyzed by DataViewer, CTan and CTvox software.
Behavioral studies
All behavioral testing procedures included the Y maze and Morris water maze, according to the methods previously described by Li et al. 22.
Luminex
Mouse serum multiplex cytokines were detected according to the instructions of the Bio-Plex Pro Human Cytokine Screening Panel, 48 Plex Kit (Bio-Rad, California, USA). Briefly, magnetic beads were added to a 96-well plate and washed with wash buffer. Mouse serum was then added to the 96-well plate with magnetic beads and incubated for 30 min at room temperature. After washing, antibodies were added, and the plates were incubated for 30 min. After washing, streptavidin PE was added, the plates were incubated for 10 min after shaking, the plates were washed, the detection solution was added, and the proteins were measured on a Bio Plex 200 system (Bio-Rad, California, USA). The lower limit of each sample for each cytokine was 50 beads. A standard curve was prepared according to the dilution standard concentration in the instructions, and the detection data were converted to pg/mL concentrations.
Quantification and statistical analysis
The Grubbs test or ROUT test was used to exclude outliers from the experimental data 23. SPSS and GraphPad Prism software were used for statistical analysis. All the data were tested for normality using the Kolmogorov‒Smirnov test or Shapiro‒Wilk test. Normally distributed data are expressed as the mean ± s.e.m., and nonnormally distributed data are expressed as the mean. The values and interquartile ranges are expressed, and the statistical analysis methods were as follows: (1) Two groups: if the data were normally distributed and consistent with homogeneity of variance, the independent sample t test was used; otherwise, the Wilcoxon signed rank test was used. (2) Paired samples: If the data were normally distributed, the paired samples t test was used; otherwise, the Wilcoxon signed rank test was used; (3) Three or more groups: if the data were normally distributed and conformed to the homogeneity of variances, then one-way analysis of variance was performed, followed by Tukey's test for multiple comparisons; otherwise, the Kruskal‒Wallis H test followed by Bonferroni correction was used for multiple comparisons. (4) Correlation analysis: If the data were normally distributed, Pearson correlation analysis was used; otherwise, Spearman correlation analysis was used. The correlation coefficient is recorded as "r", and "n=" represents the number of biological repetitions used in this study. p < 0.05 was considered to indicate statistical significance.
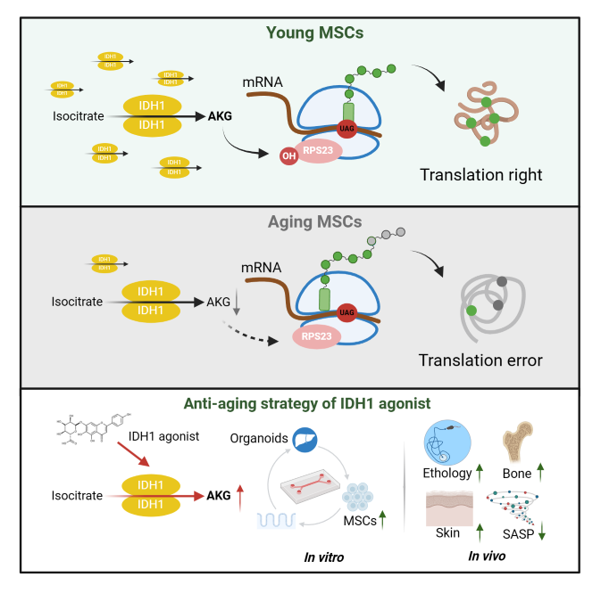{kind=link}