2.1. In silico Transcriptomic Analysis
2.1.1. Data Acquisition
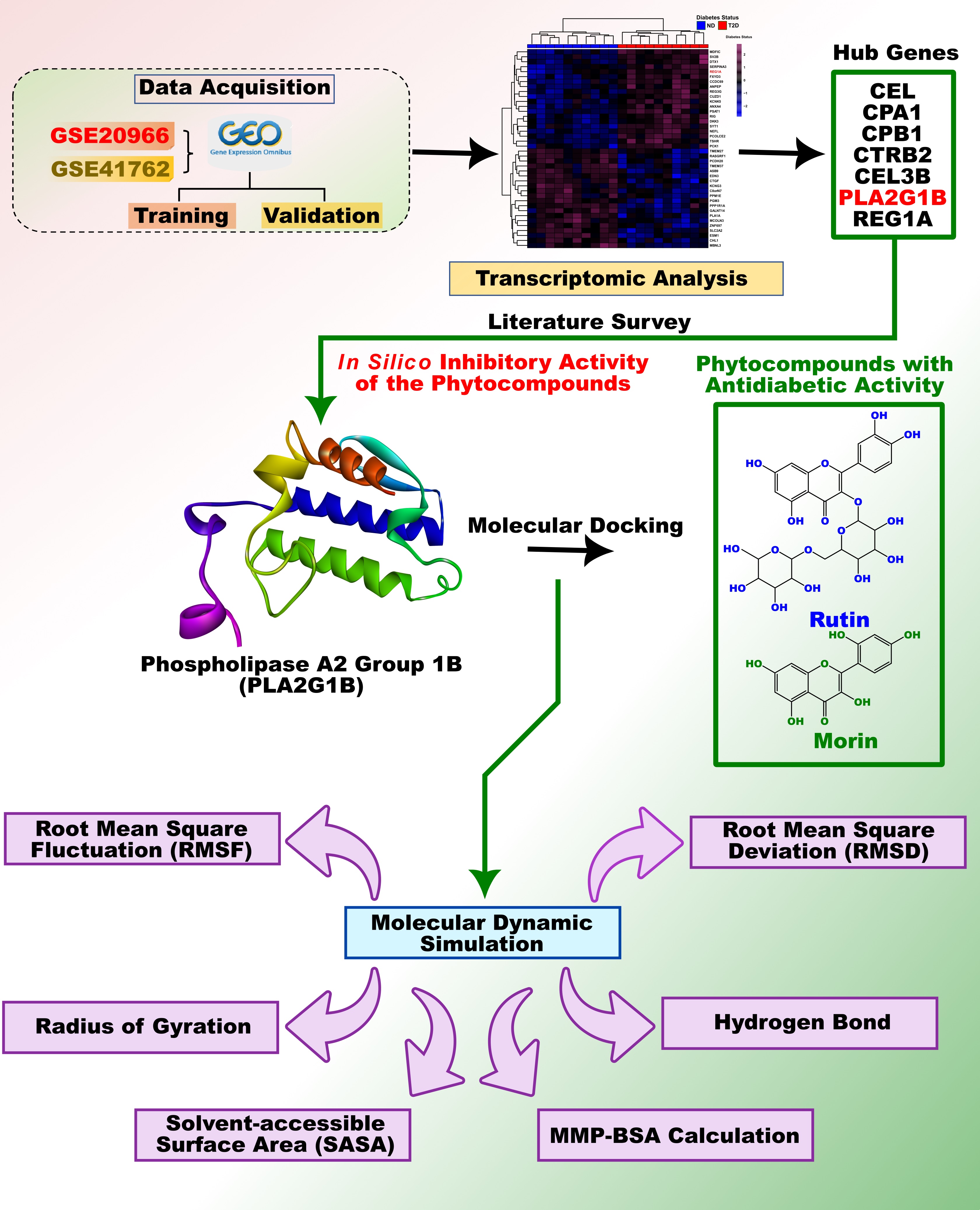
The Microarray dataset was retrieved from the Gene Expression Omnibus of National Centre for Biotechnology Information (GEO, NCBI, https://www.ncbi.nlm.nih.gov/). To gather the dataset, certain criteria were established: the dataset exclusively consisted of "Homo Sapiens", with a sample size larger than three, individuals with disease condition Type 2 Diabetes (T2D) and Non-Diabetes (ND) and lastly the dataset should specifically be focused on the “pancreas” tissue. Microarray dataset with the accession ID GSE20966 of the GPL135 platform and GSE41762 of the GPL6244 platform met the above criteria (Marselli et al. 2010; Tang et al. 2014). As per the requirement, GSE20966 encompasses 10 T2D and 10 Non-diabetics (ND) from pancreatic tissues. Likewise, GSE41762 contains 20 T2D and 57 ND patient samples (Tang et al. 2014). The raw expression matrix of both datasets along with the series matrix files was downloaded with the help of “GEOquery” package (v2.66.0) of R Bioconductor (Davis and Meltzer 2007).
2.1.2. Data pre-processing
Initially, the raw expression matrix of GSE20966 and GSE41762 have processed with Robust Multi Array (RMA) method applied from the “affy” package (v1.76.0) for background correction followed by quantile normalization and log2 transformation (Gautier et al. 2004). Probe annotation was done by converting probe IDs into gene symbol symbols with the help of the “u133x3pcdf” package (v2.18.0). In case of duplicate gene symbols with identical probe IDs, genes with the highest mean expression were selected and added to the final list of genes. For the subsequent analysis, GSE20966 was continued as a discovery dataset and GSE41762 was chosen as a validation dataset.
2.1.3. Differentially Expressed Genes (DEGs) Analysis
Normalized expression data were utilised to suspect DEGs among 10 T2D patients and 10 ND patients (T2D vs ND). The toptable function from the “limma” package (v3.54.2) (Ritchie et al. 2015) was applied to screen DEGs. This function results in a p-value, corrected p-value, log2 fold change (FC), and additional supporting parameters. Benjamini and Hochberg (BH) are one of the standardized methods employed to acquire the corrected p-value (adjusted p-value). A primary criterion of |logFC| ≥ 0.6 and a p-value < 0.05 was defined in order to screen significant upregulated and downregulated DEGs. For the visualization, a volcano plot and hierarchically clustered heatmap were created using “EnhancedVolcano” package (v1.16.0) and “pheatmap” package (v1.0.1) of R.
2.1.4. WGCNA network construction
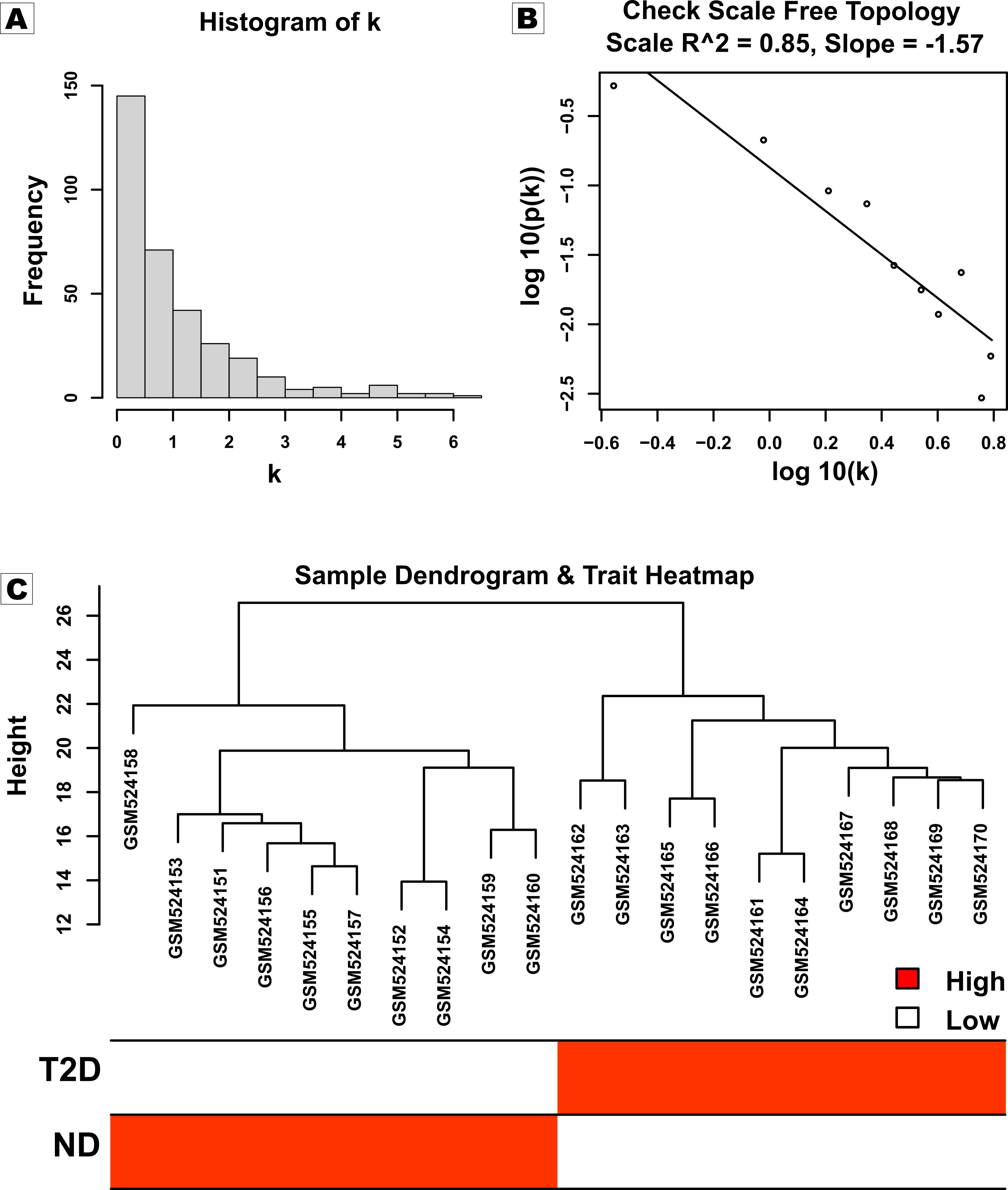
The gene expression profiles of significant upregulated and downregulated DEGs were used to construct a weighted co-expression network. Outliers were detected based on Euclidian distance by hierarchical clustering of both sample conditions. To choose an appropriate soft threshold, pick Soft Threshold function of “WGCNA” package (v1.72.1) was applied to the normalized expression matrix (Zhang and Horvath 2005). In order to achieve a scale-free network, an approximate scale-free topology was calculated with soft power β=12 (R*2=0.85, slope= -1.57). An unsigned adjacency matrix was created by choosing top strongly connected genes. Later, the adjacency matrix was converted into a topological overlap matrix (TOM), and the distance between each gene and module was determined by calculating TOM dissimilarity matrix (DissTOM). The conventional hierarchical clustering approach effectively grouped all genes into modules, each consisting of at least 20 genes. A cut height of 0.25 was employed in conjunction with the dynamic tree-cut technique to merge modules made up of genes with similar expression patterns.
2.1.5. Identification of highly correlated modules
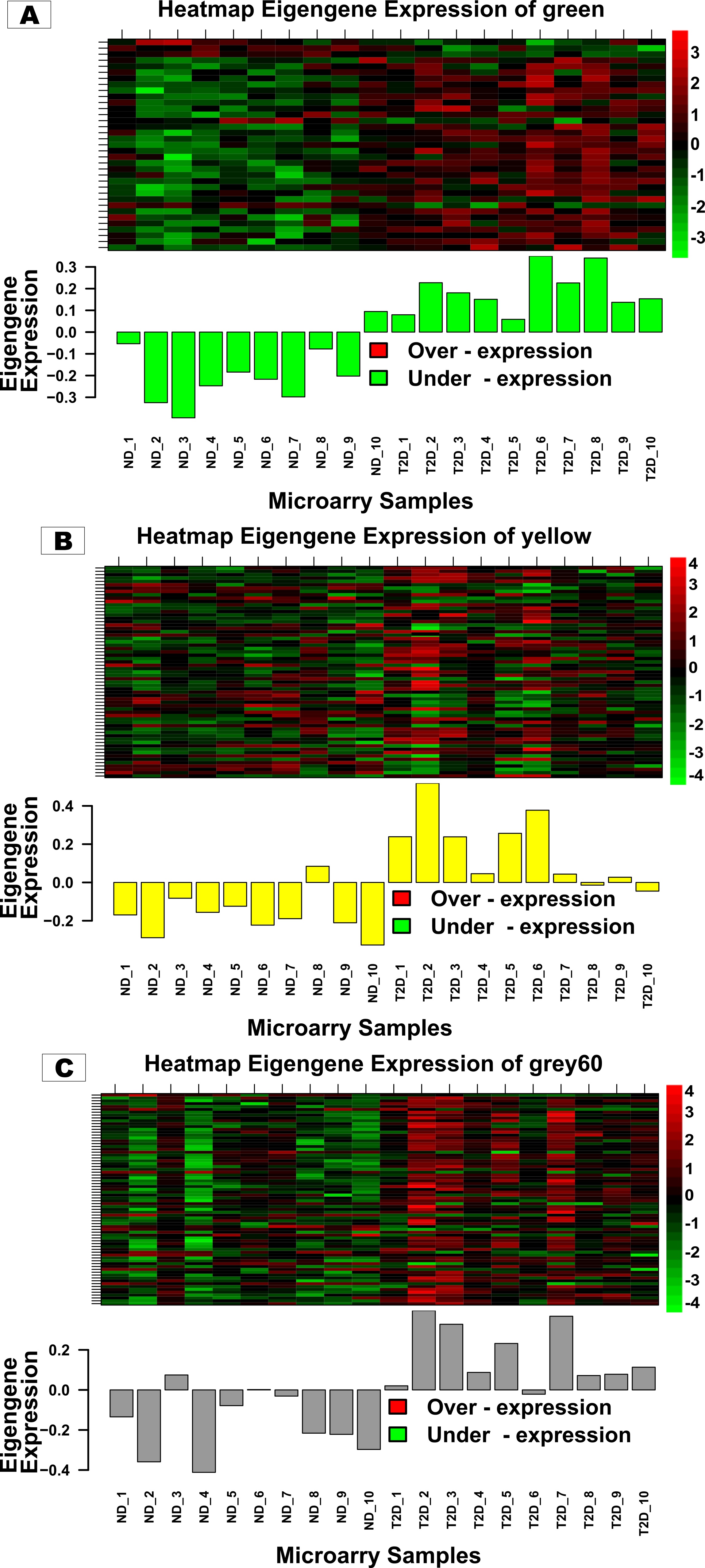
The Pearson correlation coefficient method has been employed to calculate the correlation among expression profiles with Module Eigengenes (MEs) which are termed Module Membership (MM). The above method was also applied to determine Gene Significance (GS) by calculating the correlation between each gene with T2D status as of clinical trait. Eventually, the p-value was assigned to each module-trait pair with the help of Fisher’s exact test. The value corresponds to a module-trait association close to 1 or -1 with a p-value < 0.001 is considered a highly correlated significant module.
2.1.6. Identification of hub genes network construction
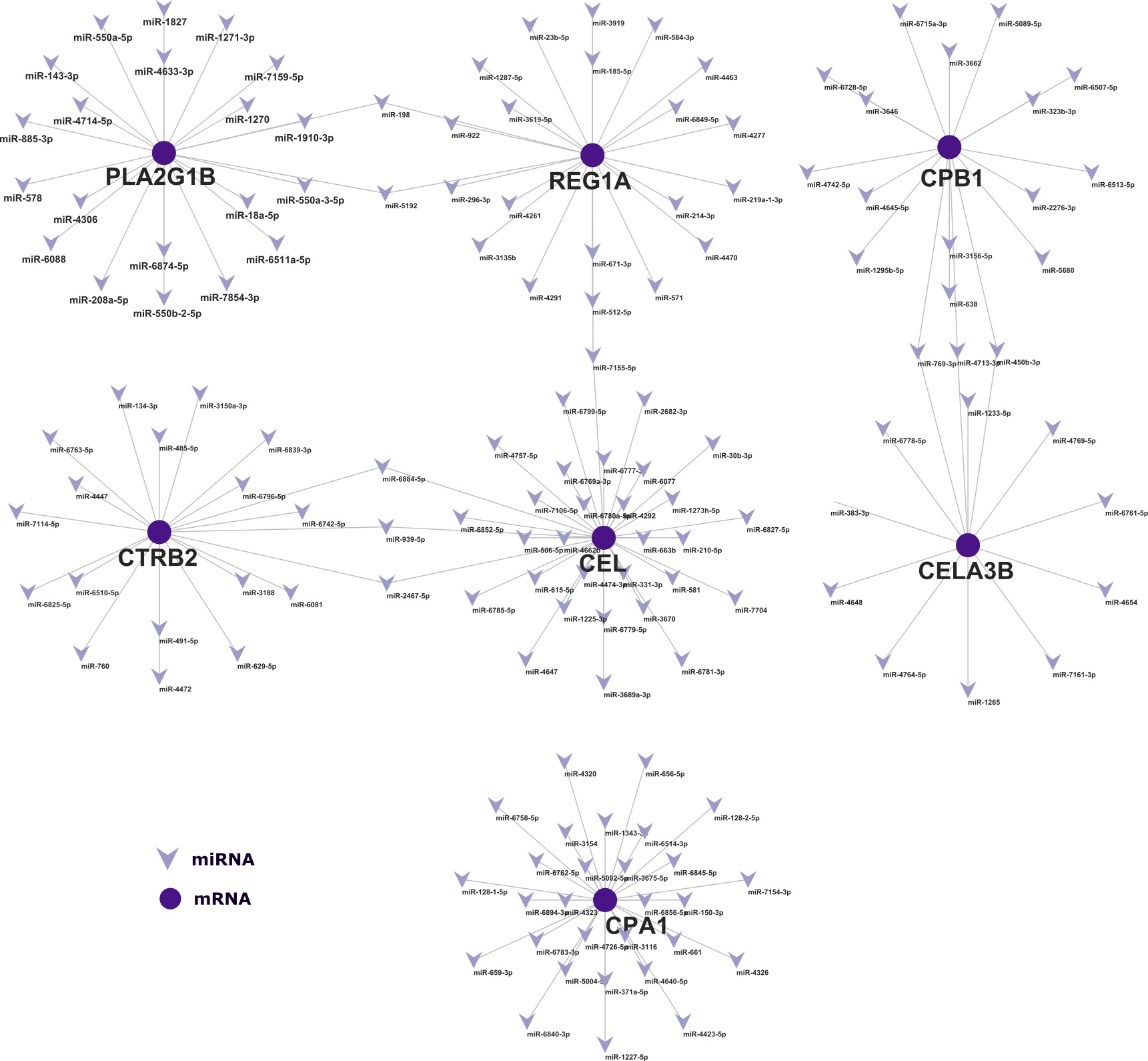
To characterize hub genes within highly correlated modules associated with T2D, MM > 0.8 and GS > 0.5 were applied as cut-off criteria. Module hub genes were uploaded to the STRING (version 11.5, https://string-db.org/) tool for the construction of a PPI network with a medium interaction score of 0.40. For the visualization and hub gene identification within the PPI network, the TSV-formatted network file from the STRING was downloaded and imported into Cytoscape (Shannon et al. 2003). Subsequently, the top genes were ranked by four methods namely MCC, MNC, Betweenness and Degree with the help of the "Cytohubba" plugin (Chin et al. 2014).
2.1.7. GO/KEGG Pathways Enrichment Analysis
To thoroughly observe the module hub genes engaged in the Gene Ontology (GO) and pathways, a comprehensive enrichment analysis was established using DAVID. The functional enrichment analysis was performed based on the Gene Ontology (GO) and Kyoto Encyclopaedia of Gene and Genome (KEGG) pathway. The enriched genes that came under GO fall into three categories such as Biological Processes (BP), Cellular Component (CC), and Molecular Function (MF). The functional terms possessing p-value < 0.05 was regarded as statistically significant enriched GO term/pathways. Lastly, all the terms and pathways were visualised with Cluego plugin of the Cytoscape and “GOplot” package (v1.0.2) of R (Bindea et al. 2009; Walter, Sánchez-Cabo, and Ricote 2015).
2.2. In silico Proteomic Analysis
2.2.1. Software and program
For primary visualization and removal of impurities PyMol version 2.5.2 (DeLano Scientific LLC, Palo Alto, California, USA) was employed. For post docking visualization assembly of the docked complex Discovery Studio Visualizer version 21.1.0.20298 (Discovery Studio Biovia 2020) (Dassault Systèmes, San Diego, California, USA) were employed. In this study, for docking, AutoDockVina version 1.2.0 (The Scripps Research Institute, La Jolla, San Diego, USA) was taken as primary docking program (Trott and Olson 2010). Open BabelGUI version 2.3.1 was used for conversion of file to the required format. The ligand.PDBQT and protein.PDBQT file format and the grid box size were prepared and determined employing AutoDock Tools version 1.5.7 (ADT; Scripps Research Institute, La Jolla, San Diego, USA).
2.2.2. Preparation of ligand structure
Chemical structures of Rutin, Morin, and n-(p-Amylcinnamoyl) anthranilic acid (ACA) were downloaded in the Spatial Data File (.SDF) file format from the PubChem Compound Database (National Center for Biotechnology Information; https://pubchem.ncbi.nlm.nih.gov/). Donor-acceptor hydrogen-bonding interactions and molecular weights are important structural factors of protein targets and ligand-binding sites (Afriza et al. 2018; Lipinski 2016; Zhang and Wilkinson 2007). Babel was used to convert the 3D structures in.SDF format to.PDB format. ADT was then utilised to analyse ligand structures in requisites of Gasteiger change additions, nonpolar hydrogen combinations and rotatable bonds. Structures in the ligand.PDB format were then converted to the ligand. PDBQT format using ADT, enabling use with AutoDock4 (AD4) and AutoDockVina (Afriza et al. 2018).
2.2.3. Preparation of protein structure
The crystal structure of the human phospholipase-A2 (6Q42) was downloaded from the RCSB protein data bank (http://www.rcsb.org). All water molecules and attached salts and inhibitors by PyMol. Further, ADT software was used to produce the necessary data for AutoDockVina by assigning hydrogen polarities, addition of Kollman charge, calculating Gasteiger charges, assigning atom type to AD4 type and converting protein structures from the .PDB file format to .PDBQT file format.
2.2.4. Possible Ligand Toxicity Prediction
The potential toxicity profiles of the ligands were studied utilising the produced simple molecular-input line entry system (SMILES) code for each chemical structure. For each candidate, the toxicity profiles included hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, toxicity class, and potential lethal dosage (LD50) value using the ProTox programme was carried out (Banerjee et al. 2018a; Banerjee et al. 2018b; Drwal et al. 2014).
2.2.5. Docking Methodology
The AutoDockVina software was used for targeted molecular docking. Ligands were docked to the receptor separately using grid coordinates (grid centre) and grid boxes of varying sizes for each enzyme and receptor. The configuration file (config.txt) was prepared from the grid box file (grid.txt) where the box center (grid center) and size (npts) coordinates are present through ADT. On the basis of the AutoDockVina scoring system, negative Gibbs free energy (ΔG) scores (kcal/mol) were projected to represent ligand-binding affinities (Trott and Olson 2010). The visualization tools such as PyMOL and Discovery Studio Biovia 2022 were used to visualise the results of post-docking analyses, which revealed the dimensions and locations of binding sites, hydrogen-bond interactions, hydrophobic interactions, and bonding distances as interaction radii of <5 Å from the position of the docked ligand. Each compound was docked to the active site or binding site of the corresponding enzyme or receptor. The best and most energetically favourable conformations of each ligand were then chosen after binding poses of each ligand were visualized and their interactions with the proteins were defined.
2.2.6. Molecular Dynamics Simulations
The GROningen MAchine for Chemical Simulations (GROMACS) 2019 version was utilized to perform molecular dynamics (MD) simulations on three complexes Phospholipase_ACA, Phospholipase_MO and Phospholipase_RU complexes (Abraham et al. 2015). In the present study utilizes GROMACS molecular dynamics software package, (Version 2019), using Charmm36-mar2019 force field for the protein parameters. The complex was solvated explicitly using the TIP3P water model inside the cubic box, and its size extends 0.1 nm away from the protein on the edges of the box in each direction. The system’s overall charge was neutralized by adding a 0.1 M salt concentration (Na+Cl−). The entire system was minimized till the maximum force was less than 10 kj/mol with the maximum steps of 50,000. The system was then equilibrated for 100ps under NVT and NPT conditions with temperature coupling for two separate groups, at 300 K. Lincs algorithm is used to constrain the bonds of the hydrogen atoms (Lemak and Balabaev 1994). Berendsen thermostat and V-rescale were used to keep the temperature and pressure constant, respectively. The cutoff distances for Coulomb and van der Waals interactions were set as 1.2 nm. Particle mesh Ewald method (PME) was used to calculate the long-range electrostatic interactions.
Then, GROMACS modules were used to do the necessary analysis, and origin was used to generate the necessary charts and figures from the 200 ns of simulations run under periodic boundary conditions (PBC). The purpose of these simulations was to ensure the flexibility and stability of the complexes that were formed after the docking process. Initially, the protein and ligand topologies were obtained separately using the 'pdb2gmx' script and the CGenFF server, respectively. These topologies were then combined to create a conformation, which underwent energy minimization using the CHARMM36 force field. The protein-ligand complex was placed in a dodecahedron and solvated with TIP3P water molecules. Counter ions were introduced to neutralize the system, followed by a rapid energy minimization using the steepest descent algorithm. The equilibration process involved restrained simulations in an ensemble with a constant number of particles, volume, and temperature (NVT), followed by another ensemble with a constant number of particles, pressure, and temperature (NPT) for 100 ps. Throughout the equilibration, thermodynamic properties such as pressure, density, potential energy, and temperature were monitored to ensure proper equilibration before the production run. The particle mesh Ewald method was employed for calculating long-range electrostatic interactions. Finally, unrestrained simulations were conducted for 200 ns at 300 K and 1 bar atmospheric pressure for each system. Simulation duration, 200 nsec could provide a more comprehensive and significant exploration of the dynamic behaviour and interactions of the protein-ligand complexes under investigation (Tantar et al. 2008). The solvated and equilibrated complexes (three protein-ligand complexes) were then subjected to the production phase to generate trajectories for root mean square deviation (RMSD), root mean square fluctuation (RMSF), hydrogen bond (Hbond) graphs, radius of gyration (RG), and solvent accessible surface area (SASA) using built-in GROMACS scripts.
The analysis like RMSD, RMSF, Rg, SASA, and Number of hydrogen bonds estimation were carried out using the Gromacs utilities. The trajectory files obtained by the MD simulation was examined using the GROMACS commands gmxrms, gmxrmsf, gmx gyrate, and gmxhbond to obtain the root-mean-square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration, and hydrogen bond interaction, respectively. After the preparation of XVG files using the Gromacsbuilit-in functions, origin was employed to plot the graph and comparing the results in both ligand-free and ligand bounded forms of the target protein.
2.2.7. MM/PBSA binding free energy calculation
To calculate the binding free energy of protein-ligand complexes, a combination of the Molecular Mechanic/Poisson-Boltzmann Surface Area (MM-PBSA) approach and molecular dynamics (MD) was employed. The final 20 ns of the final production run were specifically utilized for the calculation of binding free energy. The binding free energy estimation provides a comprehensive understanding of the interactions between the protein and ligand at a molecular level. It encompasses various energy contributions, including potential energy and solvation energies (both polar and non-polar). Further in case of MM/PBSA calculations the "MmPbSaStat.py" script was designed to automate the calculations and provide a convenient way to perform these types of binding free energy calculations for molecular complexes in a consistent and reproducible manner (Kumari et al. 2014). It takes input files containing molecular structures, force field parameters, and other necessary parameters to carry out the calculations and output the estimated binding free energies and their components. This script stands for the Molecular Mechanics, Poisson-Boltzmann Solvation Model, Surface Area, and Entropy Contributions. Molecular Mechanics involves classical force fields to compute the gas-phase energy of the molecular complex. This energy represents the energy changes due to molecular deformation and interaction in the absence of solvent effects. The Poisson-Boltzmann model calculates the electrostatic solvation energy based on the distribution of charges and dielectric properties of the molecules and the solvent. Surface Area estimates the change in solvation energy due to the exposure of the nonpolar surface area of the molecules to the solvent. This is calculated using a solvent accessible surface area model. Entropy Contributions include entropy contributions, such as the conformational entropy of the molecules, to provide a more accurate estimation of the binding free energy. The GROMACS 'g_mmpbsa' script (Kumari et al. 2014) was employed to compute the MM-PBSA binding free energies. The following equation was used to compute the binding energy in this method:
ΔG binding = G complex - (G receptor + G ligand)
The ΔG binding represents the total binding energy of the complex, while the binding energy of free receptor is G receptor, and that of unbounded ligand is represented by G ligand.
{kind=link}
{kind=link}
{kind=link}
{kind=link}