4.1. Data acquisition:
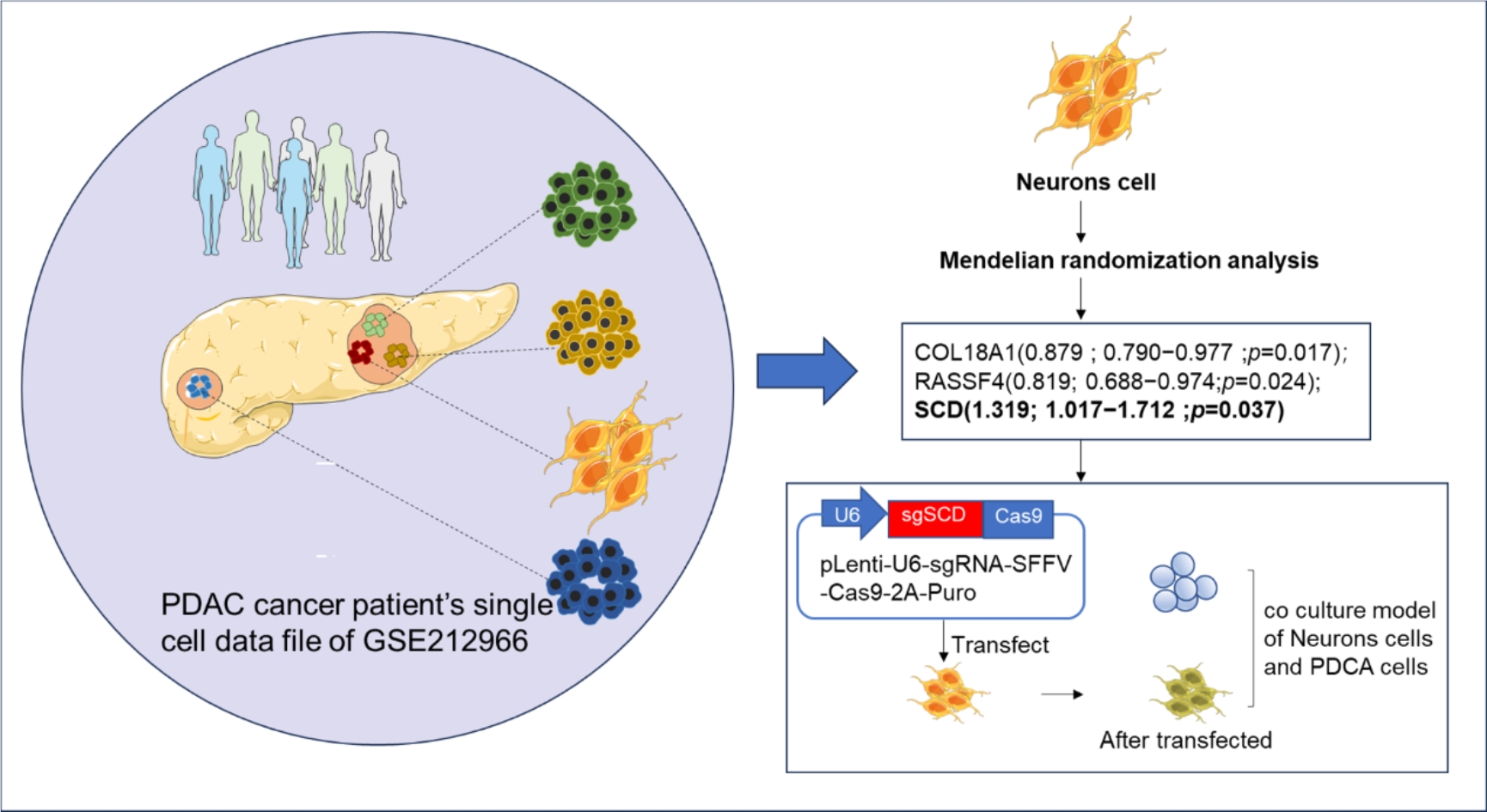
1) The GEO database (https://www.ncbi.nlm.nih.gov/geo/info/datasets.html), the full name of GENE EXPRESSION OMNIBUS, is a gene expression database created and maintained by the National Center for Biotechnology Information, NCBI. Download the single cell data file of GSE212966 from the NCBI GEO public database and include 10 sample data with complete single cell expression profiles for single cell analysis. There were 4 cases of control and 6 cases of disease.
2) TCGA database (https://portal.gdc.cancer.gov/) is currently the largest cancer gene information database, including gene expression data, miRNA expression data, lncRNA expression data, copy number variation, DNA methylation, SNP and other data. We downloaded the processed original expression data of PDAC, including normal group (n = 4) and tumor group (n = 179).
3) Exposed data: eQTL data comes from the eQTLGen consortium (https://www.eqtlgen.org) database. The eQTLGen consortium aims to study the genetic structure of blood gene expression and understand the genetic basis of complex traits. The large-scale eQTLGen project is currently in its second phase and is focused on conducting large-scale genome-wide meta-analyses in blood.
4) Outcome data: Participants in the outcome-related GWAS studies selected in this study were mainly people of European ancestry. Outcome summary data are all sourced from the EBI database (ebi-a-GCST90018893). As of now, the GWAS Catalog contains publications, top associations and complete summary statistics. GWAS Catalog data are currently mapped to Genome Assembly and dbSNP Build. Of these, there were 1,196 cases and 475,049 controls in PDAC.
4.2. Quality control:
Expression profiles were first read in via the Seurat package, where we filtered cells based on the total number of UMIs per cell, the number of genes expressed, and the percentage of mitochondrial reads per cell and the percentage of ribosomal reads per cell. Where outliers are defined as three MAD from the median. It is generally believed that cells with too high total number of UMI and expressed genes are double cells, and cells with too high mitochondrial reading percentage and ribosome reading percentage are poor quality cells that are on the verge of apoptosis or have become cell debris. After completing the above steps, use DoubletFinder (V2.0.4) to filter the double cells of each sample respectively, thus completing cell quality control.
4.3. Data standardization and cell annotation:
Use the NormalizeData function to normalize the data, use CellCycleScoring to calculate the cell cycle score, FindVariableFeatures to find hypervariable genes, ScaleData to normalize the data and eliminate the impact of mitochondrial genes, ribosomal genes, and cell cycle on subsequent analysis, and RunPCA to perform expression matrix Perform linear dimensionality reduction, select principal components for subsequent analysis, use Harmony to remove batch effects, it iteratively clusters similar cells in different batches in PCA space while maintaining the diversity of batches within each cluster, and use RunUMAP to unify Manifold approximation and projection (UMAP) performs nonlinear dimensionality reduction, FindNeighbors finds neighbor points of cells, and FindClusters divides cells into different cell clusters. By querying CellMarker and PanglaoDB databases and literature, and supplemented by automated annotation with SingleR software, we find the cell types and corresponding marker genes present in the corresponding tissue for cell annotation.
4.4. Ligand-receptor interaction analysis (Cellchat):
CellChat is a tool capable of quantitatively inferring and analyzing intercellular communication networks from single-cell data. CellChat uses network analysis and pattern recognition methods to predict the main signal inputs and outputs of cells and how these cells and signals coordinate function. In this analysis, we used the standardized single cell expression profile as the input data, and the cell subtypes obtained from the single cell analysis as the cell information. We analyzed the cell-related interactions and used the interaction strengths (weigths) between cells and The number of times (count) is used to quantify the closeness of the interaction, so as to observe the activity and influence of each type of cell in the disease.
4.5. Functional analysis of GO and KEGG:
Important genes were functionally annotated using the R package “ClusterProfiler” to comprehensively explore the functional correlation of these important genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to evaluate relevant functional categories. GO and KEGG enriched pathways with both p-value and q-value less than 0.05 were considered as significant categories.
4.6.Mendelian randomization analysis:
The EBI database contains extensive summary statistics from hundreds of GWAS studies. The outcome IDs filtered through the EBI database are extracted from the GWAS summary data (https://gwas.mrcieu.ac.uk/) and the relevant causal relationships in eQTL are selected to match the significance threshold (P < 1e-8) related SNPs as potential IVs (Instrumental variables), calculate the LD (linkage disequilibrium) between SNPs, among the SNPs with R2 < 0.001 (clumping window size = 10,000kb), only keep p2 < 5e -8 snp. In turn, it passes through Inverse variance weighted (IVW, using meta-analysis method combined with the Wald estimate of each SNP), MR Egger (based on the assumption that the instrument strength is independent of direct effects (InSIDE)), Weighted median (the weighted median method allows up to Correctly estimate causality in 50% of cases where IVs are invalid), Weighted mode (weighted model estimation has greater ability to detect causal effects, smaller bias, and lower Type I error rate than MR-Egger regression). A statistical method (if there is only one statistical method for the SNP in the causal relationship, only the Wald ratio is used) to evaluate the reliability of the causal relationship to obtain an overall estimate of the impact of all cis- and some cross-region gene expression in whole blood on PDAC.
4.7. Sensitivity analysis:
We used Mendelian Randomization (MR) leave-one-out sensitivity analysis to evaluate the impact of specific genetic variants on PDAC risk. This method identifies and eliminates variants that disproportionately affect the overall estimate by systematically excluding each SNP and recalculating the pooled effect size of the remaining SNPs. The removal of each SNP produces a new point estimate and its 95% confidence interval to assess the SNP's unique contribution and robustness to the overall results. This chart summarizes the estimates after individual SNPs are removed, as well as the overall estimate when all SNPs are included. By comparing these estimates, we can observe the impact of removing any single SNP on the overall results to determine the robustness of our analysis.
4.8.GSEA pathway enrichment analysis:
GSEA was used to further analyze the differences in signaling pathways between high and low expression groups. The background gene set is the version 7.0 annotated gene set downloaded from the MsigDB database. As an annotated gene set for subtype pathways, differential expression analysis of pathways between subtypes is performed, and significantly enriched gene sets are analyzed based on the consistency score (adjusted p value less than 0.05) to sort. GSEA analysis is often used in studies that closely combine disease classification and biological significance.
4.9. Collection of the clinical specimens
The clinical specimens involved in this study were obtained from PDAC patients who underwent pancreas at Liaoning Cancer Hospital. All patients had been pathologically confirmed and diagnosed with PDAC. Cancer tissues and matched paracancerous tissue were frozen immediately in liquid nitrogen after resection and then stored at − 80°C prior to use. The study was approved by the Ethics Committee of the Liaoning Cance Hospital and conducted following the Declaration of Helsinki.
4.10. Histology and IHC
Histology and IHC FFPE organotypic matrices and tumor tissues were processed on a Leica Peloris following standard tissue processing protocols. FFPE specimens were sectioned at 4 µm using a Leica RM2235 microtome. Sections were placed on a plain glass slide for H&E staining or a positively charged slide for IHC, which was allowed to incubate for 2 hours in a 60°C oven for maximum adhesion. Sections were deparaffinized and stained following standard H&E procedures on the Leica ST5010 Autostainer XL with hematoxylin, Australian Biostain and eosin.
4.11. Detection of mRNA expression by quantitative fluorescence polymerase chain reaction (qRT-PCR)
PCR reactions were performed with 100 ng of cDNA, using a Rotor-Gene®-Q real-time PCR cycler (Roche LightCycler 96) and TaqMan Universal PCR Master Mix (Applied Biosystems). Cycling conditions were: 10 min of denaturation at 95℃ and 40 cycles at 95℃ for 15 s and at 60℃ for 1 min. The expression levels were normalized to the expression of GAPDH, which served as the internal control. The primer sequences used for qRT-PCR are listed below:
| SDC | 5′-GCACATCAACTTCACCACATTCTTC-3′ |
| SDC | 3′-CAGCCACTCTTGTAGTTTCCATCTC-5′ |
| GAPDH | 5′-TGTGGGCATCAATGGATTTGG-3′ |
| GAPDH | 3′-ACACCATGTATTCCGGGTCAAT-5′ |
15. Immunoblotting assay
Total proteins were extracted using RIPA whole-cell lysis solution. The proteins were separated through 10% SDS-PAGE electrophoresis, measured, and semi-dry transferred to PVDF membranes (Millipore, Billerica, MA, USA). The PVDF membranes were blocked with TBS + Tween (TBST) solution containing 5% skim milk powder for 1 h, washed, and then incubated with a primary antibody (ABclonal, A16429, 1:1000) overnight at 4℃. The membranes were subsequently washed and exposed to a secondary antibody labeled with horseradish peroxidase for 1 h. Following the washing of the PVDF membrane, the chemiluminescent substrate was applied, and the grayscale values were measured using a gel imaging system.
4.12 Cell culture and transfection
Human Schwann cell line, human PDAC cell lines PCNA-1 and MIA PaCa-2 were obtained from the American Type Culture Collection (ATCC). All cell lines were maintained at 37°C in a humidified incubator with 5% CO2. In vitro growing cell lines were treated with small guide RNA (sgRNA) against SCD (ABM, China) genes and sgScramble (ABM, China). After Schwann cells were completely attached, si-RFPL4B was transfected with a final concentration of 100 nM., and incubated for 24 and 48 hours. The sgRNA sequences were listed below:
| Name | Target | Sequence |
| sgSCD-1 | 1–11 | 5′-CACATCGTCCTGCAGCAAGT-3′ |
| sgSCD-2 | 2-118 | 5′-ATGTCGTCTTCCAAGTAGAG-3′ |
| sgSCD-3 | 3-159 | 5′-TATATATGACCCCACCTACA-3′ |
4.13 Co-culture of Schwann cells and PDAC cells in vitro
Transwell-based co-cultures, 2 × 105 PDAC cells were seeded in the lower compartment of six-well plates and 2 × 105 Schwann cells were seeded on top of the Transwell membrane (Corning, 0.4 µm). After 48 h of co-culture, PDAC cells were washed by PBS three times, and then an equal number of PDAC cells were cultured individually in fresh medium for 24–48 h, then cells were harvested for further study.
4.14 Wound healing migration assay
After PDAC cells have been individually cultured for 24-48h in 6-well plate, the cells were scribed with a 200 µL tip, and the horizontal and vertical lines were scribed three times in each well. Make sure the force is uniform and the tip of the gun is perpendicular. Wash out the detached cells with preheated PBS, add 2 mL of culture medium (0–3% FBS) into each well and continue to incubate. Record at 0 hour and 48 hours respectively, and take pictures of the scratches. Image J software was used to measure the area of cell scratches, and the wound healing rate was used to reflect the cell migration ability.
4.15 Cell Proliferation Assay
After PDAC cells were cultured alone for 24h, transfected cells were plated in 96-well plates at a density of 2×103 cells/well. From the first to the third day, the serum-free medium of 10% CCK-8 (APExBIO, USA) reaction solution was used to replace the original medium and incubated for 1h. The light absorbance was quantified at 450nm (OD-450).
4.16 Statistical analysis
Reliable MR analysis is based on three premise assumptions: (1) correlation assumption (instrumental variables are closely related to exposure, but not directly related to outcomes), (2) independence assumption (instrumental variables cannot be related to confounding factors), (3) Exclusivity hypothesis (instrumental variables can only affect outcomes through exposure. When IV can affect outcomes through other pathways, it is determined that gene pleiotropy exists). In this analysis, R language (version 4.3.0) was used. All statistical tests were two-sided, and p < 0.05 was considered statistically significant. All data in this experiment were performed by GraphPad Prism 7 software. Double-tailed t-test or one-way ANOVA were used to analyzed the difference between data. The results were shown as mean ± standard error. Significant difference: *p < 0.05, **p < 0.01, ***p < 0.001.
{kind=link}