The preparation procedures of Li2S@C prelithiation agent were schematically illustrated in Fig. 1a. Specifically, the precursors of Li2SO4, polyvinylpyrrolidone (PVP), and acetylene black with a weight ratio of 4:1:1 were homogeneously mixed in the ethanol solution (solid content of 6%). After the solvent evaporation, a carbothermal reduction of the dried mixed power (depicted by the equation of 2C + Li2SO4 = 2CO2 + Li2S) was implemented under argon gas atmosphere (Supplementary Fig S1). The X-ray diffraction (XRD) pattern of the as-obtained product in Fig. 1b demonstrates the isostructural cubic phase of Li2S (PDF#23–0369). As revealed in scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 1c and Supplementary Fig. 2), the Li2S@C composite exhibits the interconnected porous network of the nanocrystalline. Accordingly, the lattice fringes with 0.33 nm d-spacing correspond to the (111) plane of Li2S, as depicted in Fig. 1d. The high-angle annular dark field (HAADF) image and elemental mapping exhibit the uniform encapsulation of sulfur species inside the thermally reduced carbon matrix (Fig. 1e). This structural feature thus enhances the electron conduction during the electrochemical-driven de-lithiation process, which in turn boosts the lithium utilization from the Li2S@C sacrificial agent. Through the precise regulation of the roll-to-roll gravure coating, it is possible to control the thickness of the slurry casting layer on the PE substrate. Correspondingly, the delithiated capacities of the Li2S@C|PE separators of varied thickness were compared in the Al||Li half cells, with the Li2S@C-coated side faced to the Al foil (details in experimental section). As illustrated in Supplementary Fig. 3, the Al||Li cell paired with the Li2S@C|PE separator (10 µm functional layer) exhibits an initial charge capacity of 1056 mAh g− 1 at 0.1 C, which reveals 91% Li+ utilization of the Li2S. With the continual increase of functional layer thickness from 5 µm to 10, 15, and 20 µm (Fig. 1f), the Li2S@C|PP separators (designated as Li2S@C|PE-5, Li2S@C|PE-10, Li2S@C|PE-15 and Li2S@C|PE-20) exhibit the gradual increase of areal prelithiation capacities from 0.24 to 0.53, 0.74 and 0.85 mAh cm− 2, respectively. Noting the Li2S@C|PP-10 achieved the highest Li+ utilization degree of 91% and optimized Gurley value of 227 s. The dimensional specifications and intrinsic physical properties of these separators were itemized in Table S1. Apparently, although the functional layers in Li2S@C|PE-15 and Li2S@C|PP-20 enhanced the theoretical prelithiation capacities, the excessive thickness adversely compromised the Gurley value (245 s and 260 s) as well as the Li+ utilization efficiency (89.5% and 86.2%). Upon immersion in the carbonate-based electrolyte, the Li2S@C|PE-5 and Li2S@C|PE-10 separators demonstrated enhanced electrolyte uptake percentages of 160% and 200%, respectively (Fig. 2g) in contrast to 127% for the pristine PE separator. This increased electrolyte absorption capability would benefit the ion diffusivity and reduce the cell's direct current internal resistance (DCIR). Meanwhile, the Li2S@C|PE-10 exhibited the highest ionic conductivity of 1.29 mS cm− 1 among the separators. Considering the harmony balance of the prelithiation capacity, Li+ utilization efficiency, and ion diffusivity, the Li2S@C|PE-10 was selected as the optimal separator, labeled as the Li2S@C|PE for the following tests. The optical image in Fig. 1h demonstrates a roll of Li2S@C|PE separator manufactured via the Roll-to-Roll gravure coating procedures (dimension of 60mm*100m), highlighting the upscaling potential for practical manufacturing. As shown in the cross-sectional SEM image (Fig. 1i), the thickness of the prelithiation layer is roughly estimated as ~ 10 µm on the PE substrate. Furthermore, the enhanced wettability and ionic percolation capability of the Li2S@C|PE-10, as compared to PE, were shown in Fig. 1j. The Li2S@C functional layer drastically reduced the contact angle (16.81°) with the carbonate electrolyte droplet on the substrate. As shown in Supplementary Fig. 4, the Li2S@C|PE demonstrated the enhanced transference number of 0.56 in the symmetric cell, which doubled the value as compared to that of the pristine PE (t+=0.33) in the Li||Li model, suggesting the facile cation permeability upon the Li2S@C functionalization.
To highlight the high-voltage tolerance and interfacial regulation effect of the prelithiation separator, the Li2S@C|PE was paired with the commercial NCM811 (2.0 mAh cm− 2) and LiCoO2 cathodes (1.8 mAh cm− 2), respectively, with reference to the Li foil. Impressively, the cycling behavior of the NCN811 that paired with the Li2S@C|PE, maintaining 94.9% capacity retention (187.3 mAh g− 1) after 200 cycles within the voltage range of 3.0-4.3 V at 0.5 C (Fig. 2a). In sharp contrast, the NCM811||Li model with the PE separator only preserved 80.8% capacity retention for 200 cycles. As displayed in Fig. 2b and Supplementary Fig. 5, the model with Li2S@C|PE also exhibits rather stable voltage profiles and much higher energy efficiency. In sharp contrast, the NCM811||Li cell with PE displayed a rapid voltage fading due to the exacerbated electrolyte decomposition upon the high-voltage charge. Moreover, the LiCoO2||Li models were assembled to evaluate even higher voltage stability within 3.0-4.5V. The cell with Li2S@C|PE separator achieved 98.1% capacity retention (182.5 mAh g− 1) without observable voltage polarization after 200 cycles (Fig. 2c-d). In comparison, the cell model with PE only exhibited 95.6% capacity retention with 155.1 mAh g− 1 maintained (Fig. 2d and Supplementary Fig. 6).
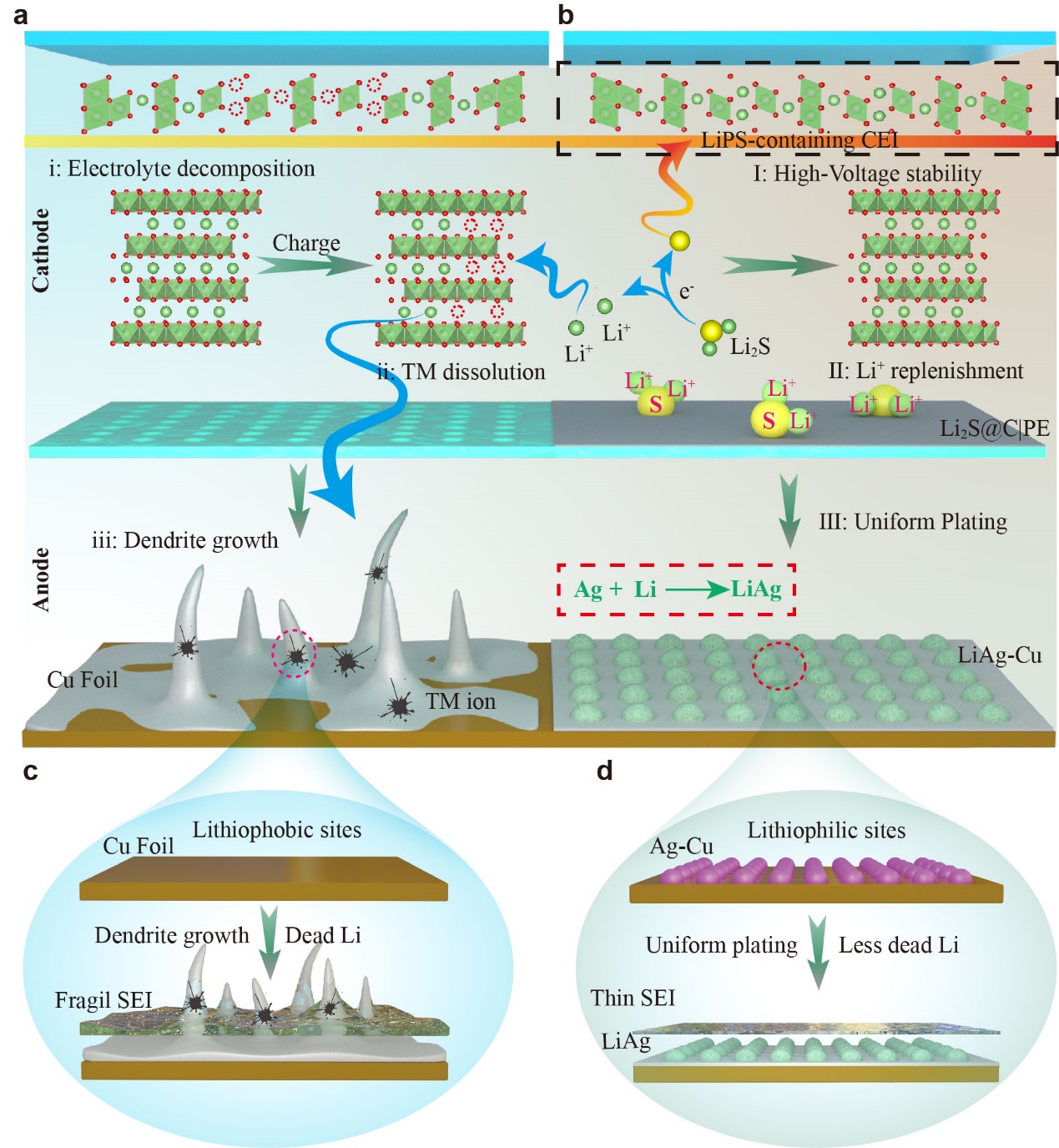
X-ray photoelectron spectroscopy (XPS) measurements were conducted to elucidate the influence of the prelithiation process on the high-voltage tolerance of the cycled NCM811 cathode. As shown in the core-level F 1s spectra (Fig. 2e), the deconvoluted sub-peaks at 684.7 and 687.2 eV could be assignable to the LixPFy and LiF species23, respectively. As compared to the PE, obviously, the interfacial species of the cycled NCM as in contact with the Li2S@C|PE separator exhibits a pronounced peak intensity of the thermal stable LiF species in the CEI layer. Moreover, the characteristic S 2p signals assignable to S-S and ROS3Li species at the binding energies of 164.3 and 169.5 eV (Fig. 2f) were observed on the cathode as in contact with the Li2S@C|PE24, 25. The S-containing species could be generally denoted as lithium polysulfides (LiPS). As further validated with morphological characterization (Fig. 2g), the post-cycled cathode with the Li2S@C|PE exhibits a conformal CEI layer with the sulfur elemental distribution at the interface (Supplementary Fig. 7), which could potentially insulate the electrolyte decomposition and TM dissolution from the NCM811. However, the non-uniform CEI layer, without the LiPS species, mainly consists of the organic-rich species derived from the solvent oxidation and contains less LiF species (Supplementary Fig. 8), these features barely prevented the NCM811 degradation at high voltage26. Moreover. the interfacial morphology of the post-cycled Li foil (Supplementary Fig. 9–10) that disassembled from the PE paired model exhibits the mossy structure. On the other hand, the Li foil from the Li2S@C|PE paired cell model was relatively smoother with less dendrite protrusion, suggesting the mitigation of TM “cross-talk” phenomenon. Figure 2h vividly illustrates the construction of LiPS containing layer on the NCM cathode. Upon the high voltage charge, the Li2S sacrificial agent could afford Li+ source upon the delithiation process, meanwhile, the sulfur species participate to form a thin and uniform CEI. Thanks to the inherent high-plasticity and electron-insulating properties, the Li-S-O and S-S species not only suppressed the TM dissolution and electrolyte decomposition, but also promoted the facile Li+ diffusion with a mitigated energy barrier. Therefore, the irreversible phasic evolution of the layered oxides was significantly restrained during high-voltage charge.
The delithiation kinetics and SEI formation evolution is investigated by the galvanostatic intermittent titration technique (GITT) and in-situ electrochemical impedance spectroscopy (EIS) during the discharge/charge process27, 28. As demonstrated in Fig. 3a-b, the thermodynamic potential of the electrochemical reactions was recorded with every period of 10 min charge/discharge and 20 min rest. The Li+ diffusion coefficient (DLi+) was also calculated to investigate the electrochemical kinetics of the cathodes (Supplementary Fig. 11). At the beginning of the charging process, the Li+ supplementary rapidly boosted the ion diffusion kinetics. The results indicate that the Li2S@C|PE significantly enhances Li+ diffusion at the NCM811 interface. More comprehensively, the in-situ EIS measurements in Fig. 3c-d reveal the in-depth mechanism of LiPS species in optimizing the stepwise charge transfer process. As reflected in typical EIS plots, three frequencies (150 kHz, 100 Hz, and 10 Hz) were selected to show the evolution of resistance with different cells. The high-frequency region (R1) is attributed to electrolyte resistance. The semi-circle frequency region (R2) could be ascribed to the interfacial resistance. The tail appearing at low frequency corresponds to a Warburg region assignable to the bulk Li+ diffusion within the electrode. All the impedance plots can be well-fitted by the equivalent circuit in Supplementary Fig. 12a. With the deeper SOC, the typical tail of the curve gradually vanishes, replaced by a depressed semicircle corresponding to the charge-transfer resistance (R3). During the charge and discharge period, the R1 was kept the constant, R2 and R3 decreased simultaneously upon charging and then increased synchronously upon discharging. Moreover, distribution of relaxation times (DRT) analysis was implemented to decouple the processes that governed the EIS spectra29, 30. As shown in Fig. 3e-f, the DRT plots of the EIS after the discharging process shows six peaks within a timescale (τ) range of 10− 6-10− 1 s, denoted as τ1–τ6. To reveal the interface chemistry with Li2S@C|PE separator, we assigned these DRT peaks to specific processes and tracked their evolution during the first cycle. Specifically, contact resistance (τ1), ion transport through the CEI layer (τ2–τ3), charge transfer (τ4–τ5), and mass transport (τ6) were assigned, respectively. As shown in Fig. 3E and S13, charge transfer resistance at the interface (τ4–τ5) dominates upon whole process, while the value of τ5 increases sharply after the complete cycle. This indicates high charge resistance between NCM811 and electrolyte, severely hindering the ion diffusion in the cathode. In comparison, the NCM811||Li with Li2S@C|PE separator preserved relatively lower charge resistance (Fig. 3f and Supplementary Fig. 14). Noting that the difference in the τ4 peak is due to the varied Li deficiency in the structural lattice of NCM811, which leads to different Li diffusivities. Meanwhile, the τ2 and τ3 peaks could be assigned to the ionic transport at the interfacial CEI. Notably, the peak of the model with Li2S@C|PE separator hardly increased after the initial CEI film formation, whereas the cell with PE separator showed the pronounced charge transfer resistance. As illustrated in Supplementary Fig. 12b, the interfacial resistance (R2, 15.4 Ω) of NCM811||Li paired with Li2S@C|PE were maintained much lower as compared to that with PE (47.6 Ω), mainly owning to the interfacial stabilization effect after the introduction of Li2S@C layer. In addition, the charge-transfer R3 values for the Li2S@C|PE equipped cells (38.8 Ω) are smaller than the counterpart with PE (118 Ω). These results suggest the kinetics activation effect through pairing the Li2S@C|PP separator, which favors the Li+ migration across the multiple interphases in the NCM811||Li model.
As a proof-of-concept to evaluate the prelithiation effect, the models were assembled by pairing the NCM811 cathode, bared Cu foil with either Li2S@C|PE or PE separators. For the AFLMB architecture, there exists no cation reservoir at the anode part, which thus allows for the evaluation of Li+ compensation effect provided by the Li2S@C agent for the cell. Meanwhile, the interfacial incompatibility between carbonate electrolyte and Li deposits on the Cu foil would aggravate the electrolyte decomposition and irreversible Li+ depletion on the substrate. As compared in Fig. 4a-c, the NCM811||Cu model with Li2S@C|PE delivered an initial reversible capacity of 194.7 mAh g− 1 and moderate capacity retention of ~ 60% for 50 cycles. In sharp contrast, the NCM811||Cu with PE cell experienced rather severe capacity fading after only 10 cycles. Accordingly, the average Li inventory retention rates (LIRR) were calculated as 99.1% and 85.0% for the NCM811||Cu models with Li2S@C|PE or with the PE, respectively, which could be utilized as reasonable metrics to assess the “average” Li inventory loss per cycle. Therefore, the Li extracted from Li2S@C|PE indeed offset the irreversible Li inventory and alleviated the capacity fading. Meanwhile, the transmission-mode operando XRD was employed to probe the multi-phase evolution, more importantly, the dynamic interplay between the supplement cation inventory and the cathode31. As revealed in the contour plot (Fig. 4d-i), the (003), (101), (104), (105), and (107) diffraction planes of the NCM811 cathode were clearly observed. The hexagonal (003) plane at 8.63° represents the c-axis lattice breathing of the layered oxide lattice. During the initial charge process, accompanied by the gradual disappearance of the Li2S peak, the characteristic (003) peak of NCM811 begins to shift towards the lower 2θ angles. This process could be assigned to the phasic evolution from the H1 to H2 and corresponding lattice expansion. Upon the subsequent charging, the peak quickly shifts to the higher angles, indicating the subsequent transition to H3 phase32. To quantitatively assess the Li+ reversibility of the cathode framework, the peak shift of (101) planes was employed to calculate the variation of the a-axis. According to lattice parameters shown in Fig. 4f, the c value is similar to the curve trend of the (003) planes as the Li+ source extracted from the NCM811 cathode. The variations of a and c lattice parameters of the NCM811 that paired with PE were estimated as 1.9% and 2%, which indicates lithium depletion leads to the Li deficiency of the oxide lattice. In comparison, the cell model with the Li2S@C|PE exhibits only 0.3% and 0.17% variations of a and c lattice after a complete cycle (Fig. 4i). These results demonstrate that the supplementary lithium source could alleviate the lattice distortion of the NCM. Meanwhile, the diffraction peaks that ascribed to the Li2S gradually evolved into the sulfur precipitation upon the initial charge process (Fig. 4h), which echoed with the S-S containing species observed on the cathode (Fig. 2f).
The reversible Li plating/stripping behavior on the bared substrates hinges on the initial nucleation process, which overwhelmingly affects cycling endurance and operational safety of AFLMB models. Owning to the intrinsic lattice mismatch with metallic lithium, the copper foil exhibits lithiophobicity and thus the high nucleation overpotential33. Therefore, a modification layer of 100 nm Ag nanoparticles (Ag NPs) was purposely deposited onto the Cu substrate, denoted as Ag-Cu. The top-view SEM images of the pristine Cu foil at various magnifications revealed a smooth and flat surface, in contrast to the grey-color, rough surface of Ag-Cu foil with interfacial particles (Fig. 5a-b). The XRD pattern confirms the presence of cubic metallic Ag with the diffraction peaks situated at 17.2° (Supplementary Fig. 15). Notably, the coating thickness did not vary significantly as compared to 6 µm Cu foil (Supplementary Fig. 16). Energy-dispersive X-ray spectroscopy (EDS) cross-sectional mapping further verified the compact, uniform coverage of Ag NPs film on the Cu foil. The Ag 3d spectra can be deconvoluted into dual subpeaks located at 367.9 eV and 373.9 eV (Ag 3d5/2 and Ag 3d3/2), suggesting the presence of zero-valent Ag (Fig. 5c). Supplementary Fig. 16 demonstrates the lightweight (0.1 mg cm− 2) layer with nano-thickness. This coating layer thus has not enhanced the weight/volume of the Cu substrate too much. The EIS measurements, combined with the equivalent circuit simulation (Supplementary Fig. 17), suggest the Li||Ag-Cu model demonstrated a much lower interfacial resistance (22.5 Ω) as compared to the Li||Cu counterpart (35.3 Ω). Based on the analysis of the Li-Ag phase diagram (Supplementary Fig. 18), the formation of a series of Li-Ag alloy intermediates (LiAg, Li9Ag4, and Li10Ag3 phases) could reduce the nucleation barrier upon the pronounced Li solubility 34, 35. Consequently, the Ag-Cu anode demonstrates a higher initial Coulombic efficiency (ICE) of 92.9% for the initial plating/stripping process, in contrast to 84.5% for the Cu substrate at 1 mAh cm− 2 (Supplementary Fig. 19). Corresponding to the plating curve of the Ag-Cu substrate (Fig. 5d), the transmission-mode XRD characterization tracked the stepwise lithiation alloying process in operando, validating the gradual formation of LiAg with the Li+ solubility amount up to 0.04 mA cm− 2, Li9Ag4 with 0.062 mA cm− 2 and Li10Ag3 with 0.097 mA cm− 2 Li contents, respectively. Based on the Aurbach CE calculation methods36, the average CE of Li plating/stripping on Ag-Cu could reach the value of 99.2% from the second cycle onwards, significantly higher than the 97.9% on the Cu foil (Fig. 5e).
As scrutinized in the post-cycled SEM images (Supplementary Fig. 20–21), the Ag-Cu substrate retained less “dead Li” residue than that on the Cu foil, indicating higher Li utilization efficiency. In-situ optical microscopy was employed to visualize the Li deposition process on both substrates. Figure 5f exhibits whisker-shaped lithium deposits that appeared on the Cu anode after 10 mins of plating, which further protruded into the dendrites after 60 mins. In sharp contrast, the Ag-Cu substrate (Fig. 5g) maintains a smooth surface without obvious dendritic morphology, highlighting the beneficial Li-Ag alloy layer to favor the homogeneous nucleation. The schematic diagrams in Fig. 5h-i illustrate the lithium deposition process on both Cu and Ag-Cu substrates. During the initial charge, the lithiophobic Cu foil with a higher nucleation barrier tends to induce the localized charge transfer process at the nucleates, which easily accumulates into the sharp spike. However, the ultrathin Li-Ag intermediate alloy enhances lithium affinity and high-throughput Li+ influx, enabling the homogeneous Li electrodeposition. Quantitative numerical estimations of the accessible lithium source were further explored. The irreversible Li trapping in the intermediate alloy formation only consume 0.097 mAh cm− 2 during the first cycle, which could be sufficiently counterbalanced by the prelithiation reservoir (Li2S@C, 0.5 mAh cm− 2) from the separator. Therefore, this cell prototyping could avoid the cathode inventory depletion during the initial formation cycle, guaranteeing the retrievable capacity of the cell.
To evaluate the performance metrics of practical relevance, various NCM811||Cu, NCM811||Ag-Cu models paired with Li2S@C|PE separator, in the formats of the coin cells and pouch cells, were assembled under the lean electrolyte conditions (LiFSI-1.2DME-3TTE, 10 µl mAh− 1). So far, seldom studies could balance the robust cycling endurance and the extreme energy densities for the AFLMB models at the Ah-level prototyping, in terms of the Wh kg− 1 or Wh L− 1 values. As shown in Fig. 6a, the NCM811||Cu cell with PE separator continuously decayed and only maintained the 40.2% capacity retention for 100 cycles, in comparison, the NCM811||Ag-Cu model with PE separator preserved 72.0% capacity retention. Noting that the NCM811||Ag-Cu cell with Li2S@C|PE separator exhibits the superior cycling endurance (87.4% capacity retention for 100 cycles), which further validates the synergistic coupling of the Li2S@C|PE separator and substrate modification. Moreover, the NCM811||Ag-Cu model with the Li2S@C|PE exhibits an apparent alloying plateau, corresponding to the activation of Li-Ag alloy formation (Fig. 6b). The following discharge/charge cycles maintained cycling endurance without any overpotential fluctuation or voltage decay in the curves (Supplementary Fig. 22). To assess the upscaling potential of the prelithiation strategy, the 1.22 Ah, layered-staked AFLMB model was assembled by pairing the NCM811 cathode (double sided, 25 mg cm− 2 for each side), and Li2S@C|PE separator, as well as the Ag-Cu substrate (Fig. 6c). The technological specifications of the pouch cell were itemized in Fig. 6d. The NCM811||Ag-Cu model with the Li2S@C|PE exhibits long-term cycling endurance and maintains 85% capacity retention after 100 cycles (Fig. 6e); by contrast, the NCM811||Cu model paired with PE suffered from severe capacity fading upon the cycling (only 32% capacity retention for 100 cycles). This comparison validates the effective Li+ supplementary from prelithiation separator strategy. Furthermore, superior rate capabilities of the NCM811||Ag-Cu cell with Li2S@C|PE were demonstrated, with reversible capacities of 205.2, 190.2, 172.3, 139.6 and 113.7 mAh g− 1 obtained at 0.1, 0.5, 1, 2, and 3C, respectively (Fig. 6f). Figure 6g demonstrates the Ragone plot of the as-assembled pouch cell (based on both the total mass of the whole device. The NCM811||Ag-Cu model with the Li2S@C|PE exhibits a practical gravimetric/volumetric energy densities of 450 Wh kg− 1/1355 Wh L− 1 at the cell level, far surpassing the previously reported anode-less/free based battery configurations from the literature. If the calculation was based on the total mass of the electroactive materials, a superior energy density of 779 Wh kg− 1 could be obtained. Besides, the extreme power output of 830.6 W kg− 1 could be achieved for the cell model (Table S2). Owning to the multiscale interfacial therapy from the Li2S@C|PE separator with the additional Li+ inventory, the AFLMB prototypes overcome Li+ consumption origins and boost the cation transfer kinetics, especially as applications move toward energy/power-dense regimes.
{kind=link}