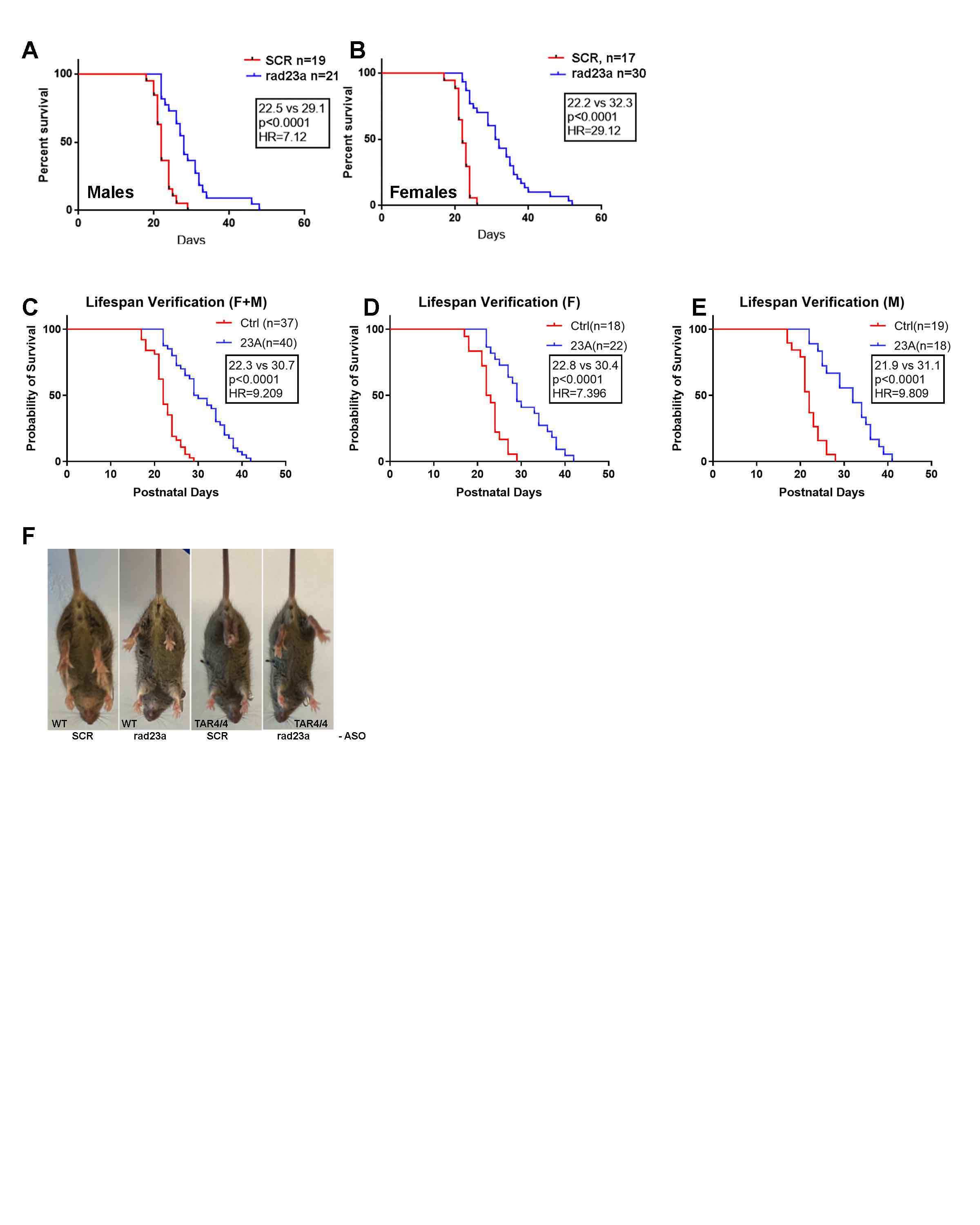
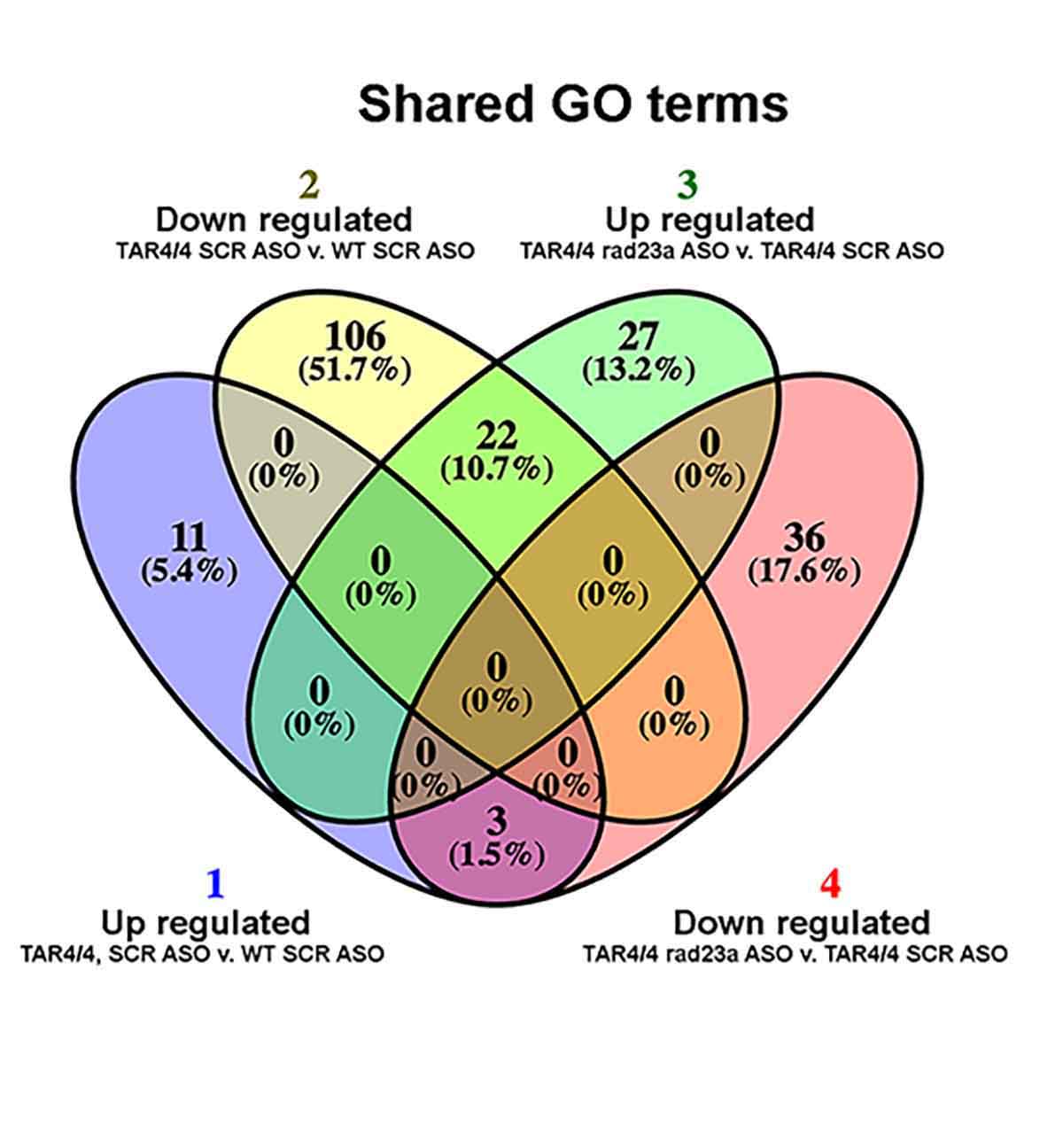
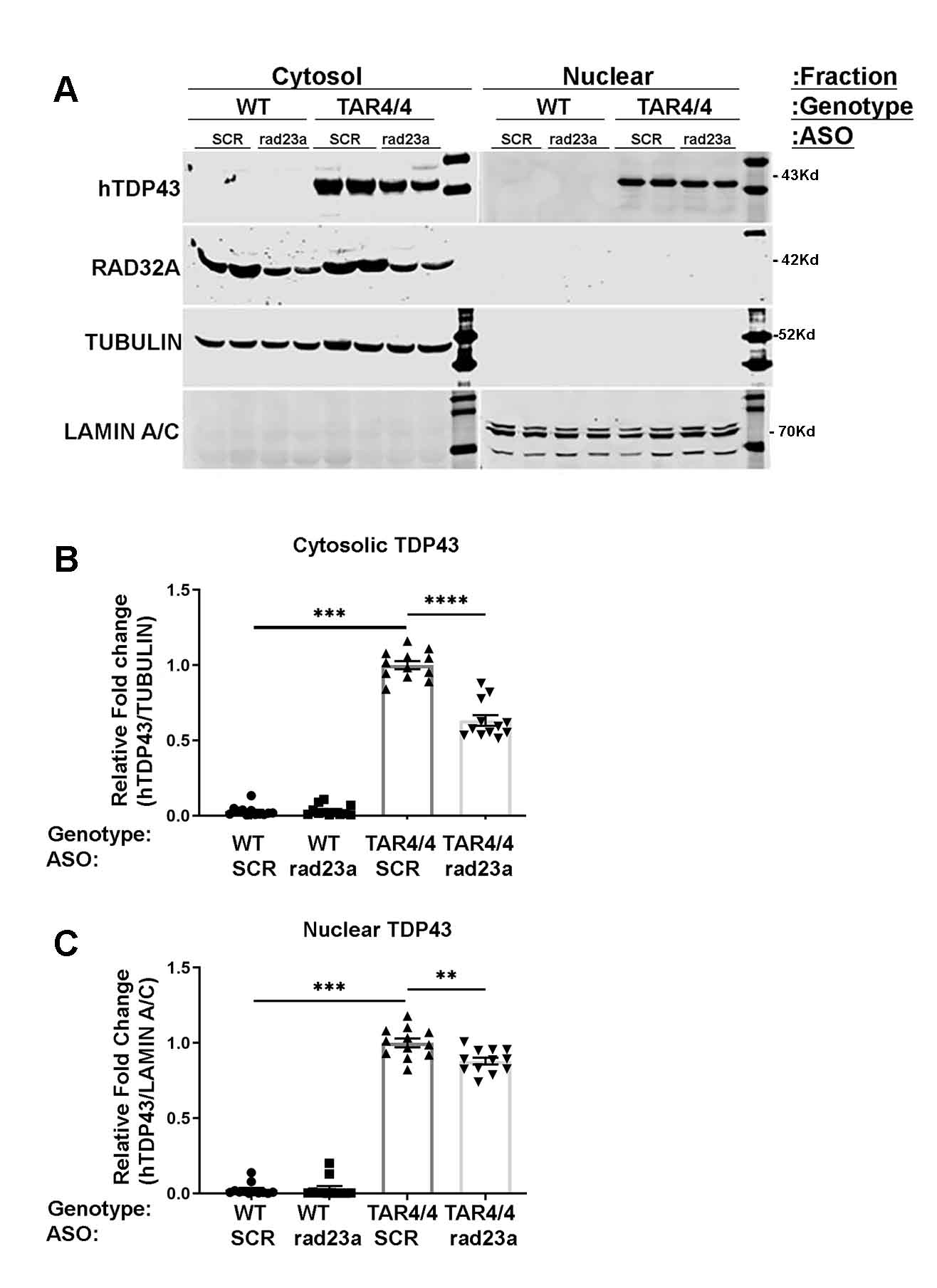
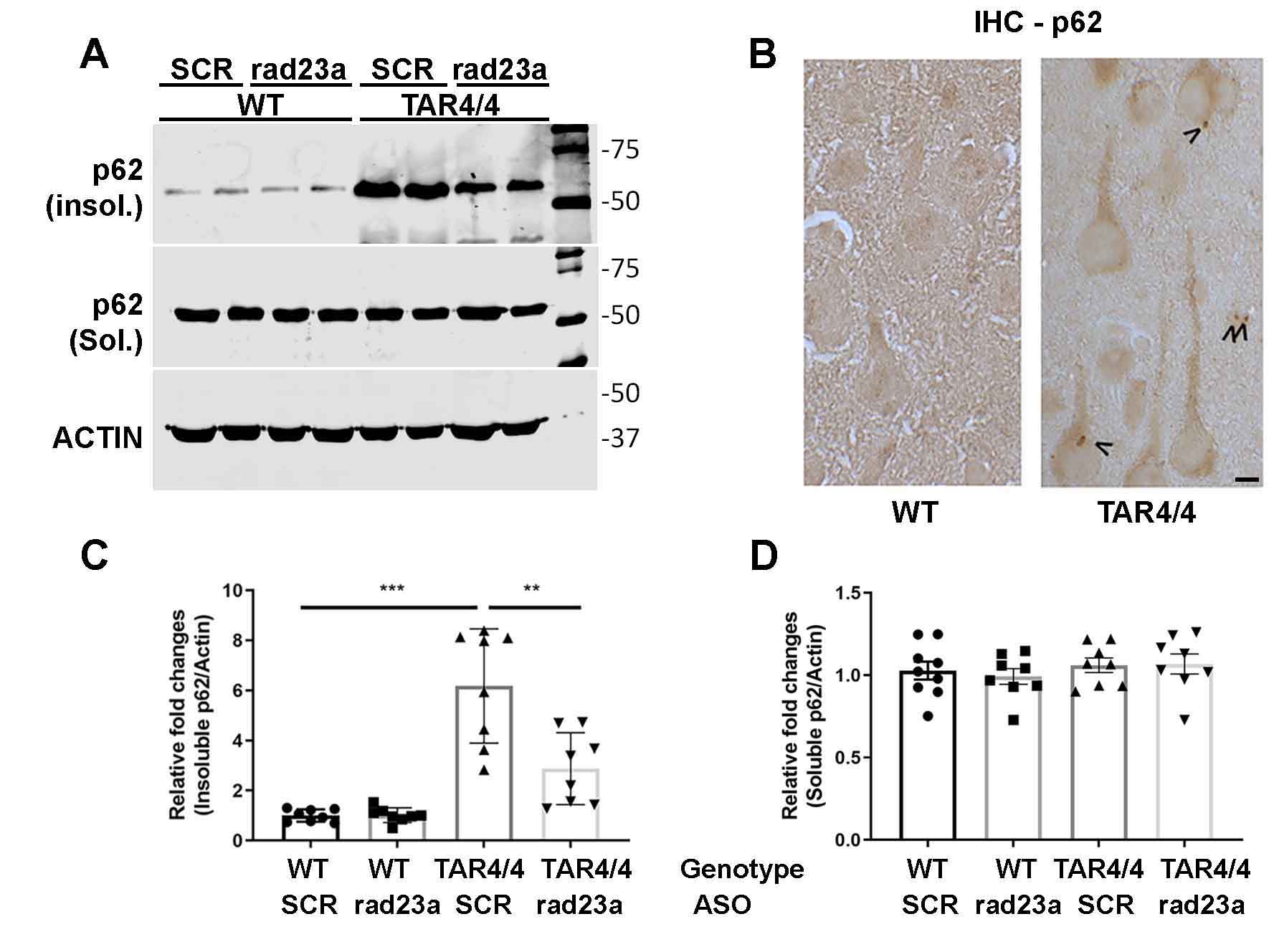
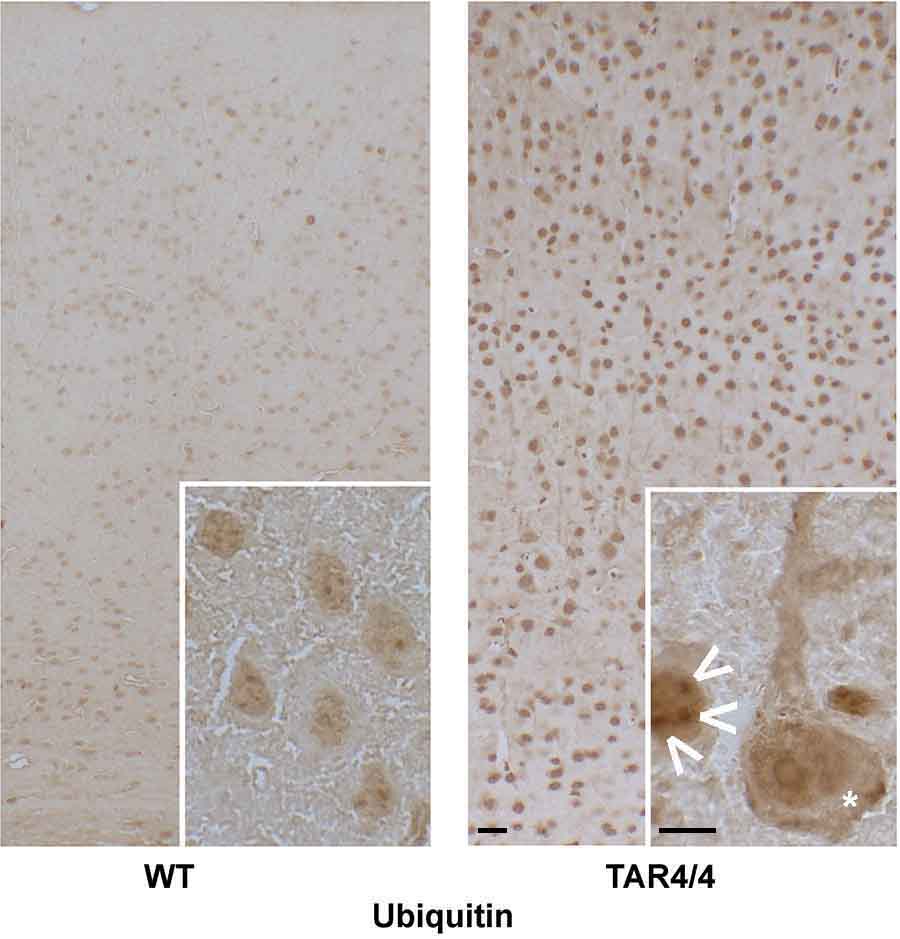
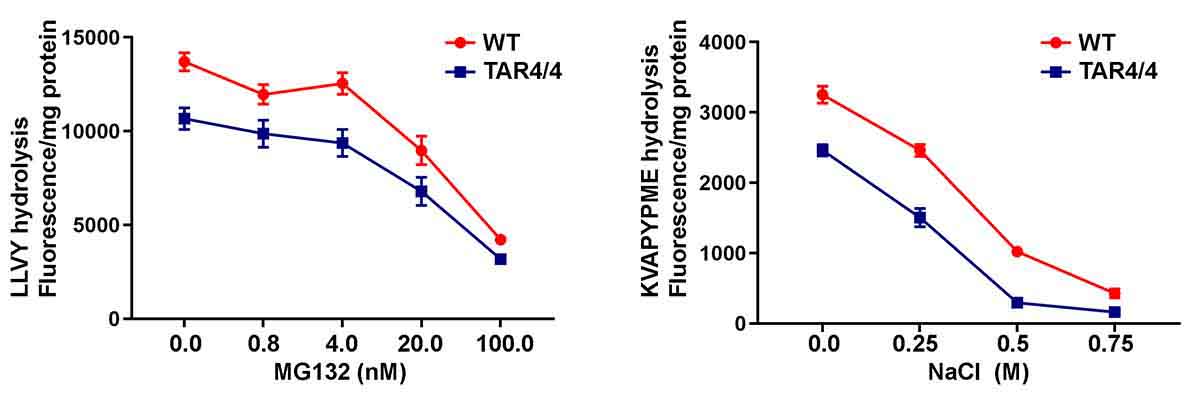
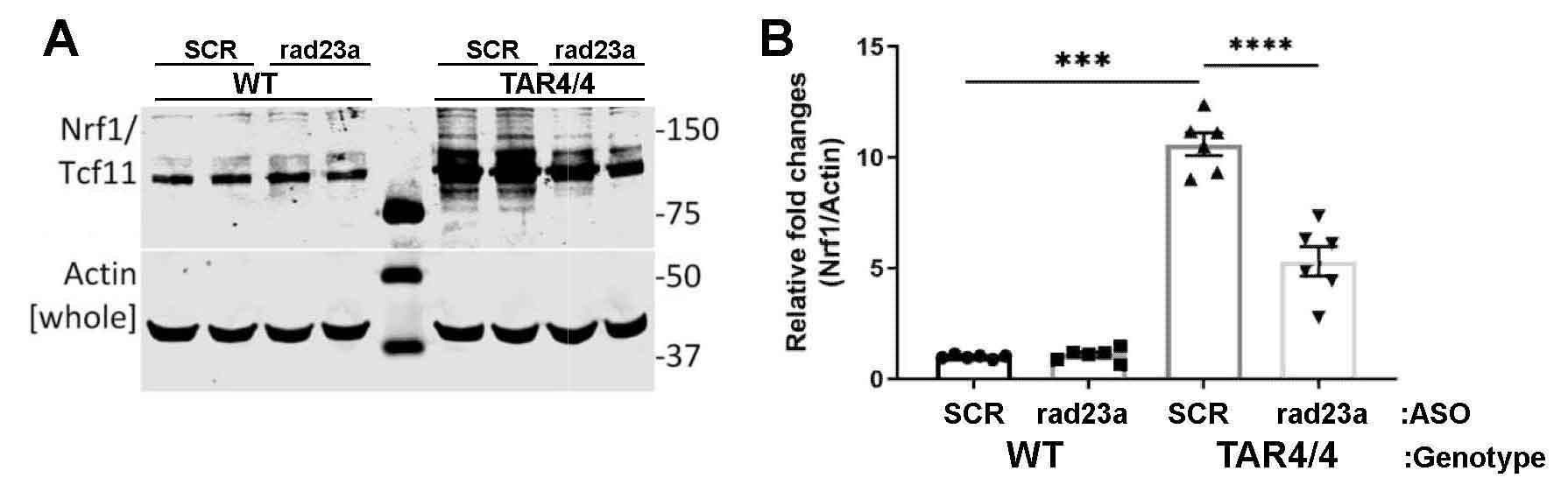
Protein misfolding and aggregation are cardinal features of neurodegenerative disease (NDD) and they contribute to pathophysiology by both loss-of-functon(LOF) and gain-of-functon(GOF) mechanisms. This is well exemplified by TDP-43 which aggregates and mislocalizes in several NDDs. The depletion of nuclear TDP-43 leads to reduction in its normal function in RNA metabolism and the cytoplasmic accumulation of TDP-43 leads to aberrant protein homeostasis. A modifier screen found that loss of rad23 suppressed TDP-43 pathology in invertebrate and tissue culture models. Here we show in a mouse model of TDP-43 pathology that genetic or antisense oligonucleotide (ASO)-mediated reduction in rad23a confers benefits on survival and behavior, histological hallmarks of disease and reduction of mislocalized and aggregated TDP-43. This results in improved function of the ubiquitin-proteasome system (UPS) and correction of transcriptomic alterations evoked by pathologic TDP-43. RAD23A-dependent remodeling of the insoluble proteome appears to be a key event driving pathology in this model. As TDP-43 pathology is prevalent in both familial and sporadic NDD, targeting RAD23A may have therapeutic potential.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}