Characterization of photocatalysts
The in situ growth of 2D In-MOF on single-layered GO was triggered by separately injecting aqueous solutions of In(NO3)3 and tetrakis(4-carboxylpheny) porphyrin (TCPP), the precursors of In-MOF, into a GO suspension dispersed in a DMF/EtOH mixture (V/V = 3/1) (Fig. 1a). The injection rates of both precursor solutions were precisely controlled at a rate of 0.25 mLཥh− 1 using an injection pump. The growth procedure was performed for 6 h; samples were collected hourly for monitoring using transmission electron microscopy (TEM) and X-ray diffraction (XRD). The samples were named In-MOF/GO-xh, where x denotes the hour after which the sample was taken. The pristine GO sheet (In-MOF/GO-0h) has a clean surface with a typical layered structure (Fig. 1b), characterized by a single XRD peak at 11.4° corresponding to the (100) plane25 (Fig. 1c). The absence of additional peaks in the XRD patterns (Fig. 1c; In-MOF/GO-0h and In-MOF/GO-2h) indicates that during the first 2 h of in situ growth only small In-MOF nuclei, which have not crystallized, are present on the GO sheet (Fig. 1b). Prolonged growth for more than 3 h results in the formation of a sheet-like 2D In-MOF with its characteristic XRD peak at 7.5° assigned to the (021) plane of In-MOF26,27. After 4 h of growth, cubic nanosheets of In-MOF are clearly observed to be well-dispersed on the GO surface in a face-to-face arrangement, forming a well-defined 2D/2D heterojunction. In Fig. 1c, the well-resolved (021) peak in the XRD pattern of In-MOF/GO-4h demonstrates full crystallization of the 2D In-MOF. After 6 h of elapsed growth time, the GO surface becomes fully occupied by In-MOF nanosheets, aggregated in random orientations (Fig. 1b; In-MOF/GO-6h). Notably, the synthetic approach for In-MOF nanosheets presented in this study is a modified version of a previously reported procedure26. The XRD patterns (Fig. 1c) of the as-synthesized In-MOF nanosheets match well with those of the previously reported In-MOF (Supplementary Fig. 1b). As shown in Supplementary Figs. 1c and d, the previously reported In-MOF contains In-nodes coordinated with four oxygen atoms from the basal plane ligands and two oxygen atoms from the axial OH groups, forming InO4(OH)2 chains26.
The In-MOF/GO-4h sample, featuring well-dispersed In-MOF nanosheets without aggregation, was further characterized. High-resolution TEM (HR-TEM) analysis reveals a lattice fringe of 1.153 nm (Fig. 1d), corresponding to the (021) plane of In-MOF26 (Supplementary Fig. 2). High-angle annular dark-field (HAADF) mapping showed that the 2D In-MOF formed a face-to-face dominant architecture on the GO sheet (Fig. 1e). Atomic force microscopy (AFM) was used to estimate the thickness of the 2D In-MOF.The thickness of the in-situ grown In-MOF nanosheets is determined to be approximately 0.8 nm (Supplementary Fig. 3).
Photocatalytic activity test
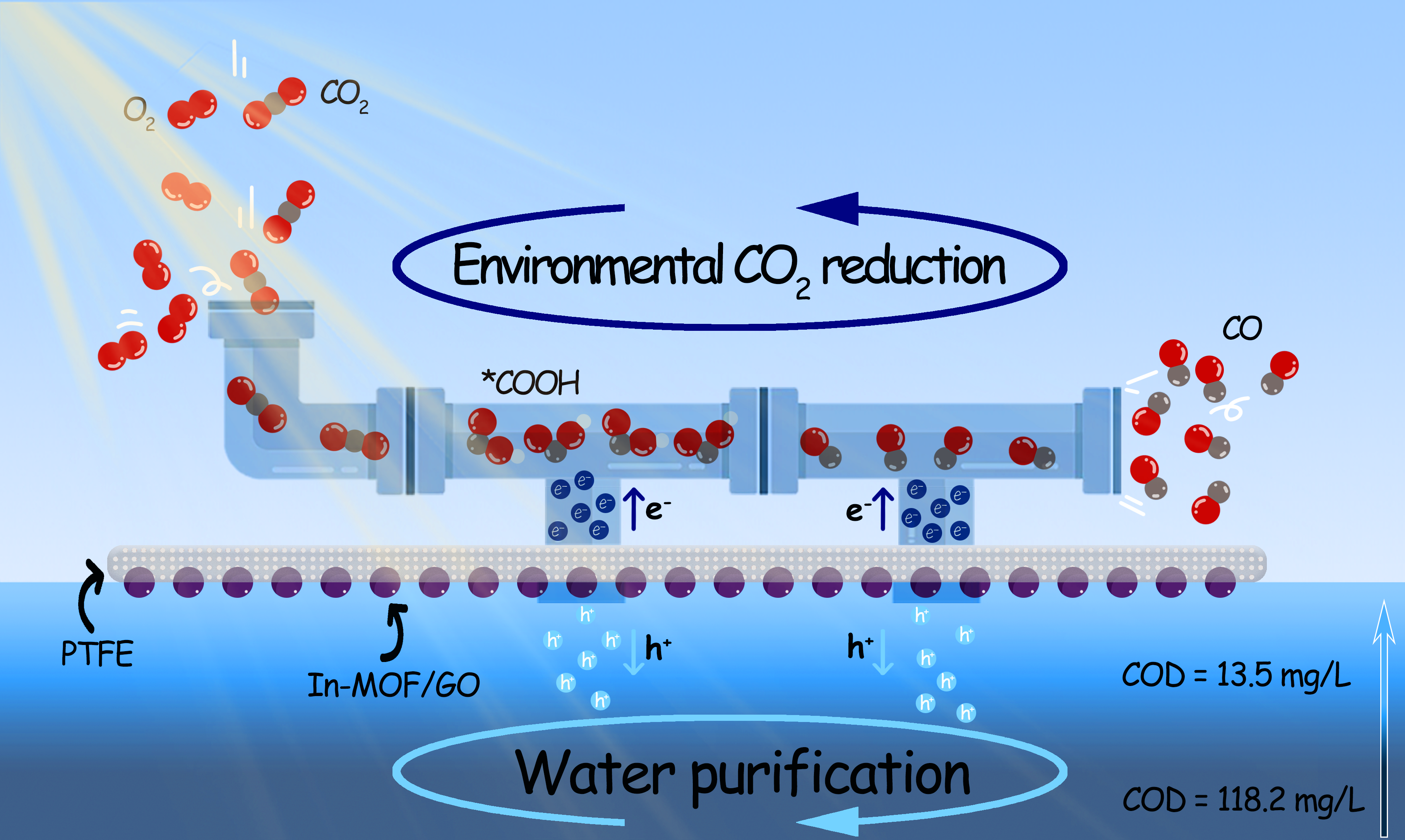
The as-synthesized In-MOF/GO was further integrated into the hydrophobic pore structure of a PTFE membrane via vacuum filtration to construct an artificial leaf. This artificial leaf could float on water and initiate a triphase photocatalytic process (Fig. 2a). After orienting the In-MOF/GO catalyst-embedded side of the artificial leaf downward and the other side with an open pore structure upward, the hydrophobic pore structure of the integrated system can function as a gas-diffusion layer to facilitate the transportation of gaseous reagents to the embedded catalysts. Integration of MOF into a floatable device rather than simply dispersing it in an aqueous phase enhances the effect of the MOF pore structure in enriching gaseous reagents. The limited solubility of CO2 in water renders floatable photocatalytic devices beneficial for reactions involving sources with low CO2 concentrations. The triphase photocatalysis was first conducted in deionized water with 10% CO2 (mimicking typical CO2 levels in combustion flue gases), 0–20% O2, and 90–70% Ar in the gas phase. Even in the presence of air-level O2 (20%), the artificial leaf demonstrated a high CO generation rate of 762.5 µmolཥg− 1ཥh− 1 with 100% selectivity, without notable H2 generation28. The performance demonstrated by the constructed artificial leaf under aerobic conditions surpasses the performances of most reported MOF-based and inorganic catalysts under anaerobic conditions (Supplementary Table 1).
For photocatalytic CO2 reduction, the use of artificial leaves loaded with In-MOF/GO-4h exhibits 20.3 and 32.9 times higher CO generation rates than those of In-MOF or GO, respectively (Fig. 2b). It is worth noting the aim of in-situ growth approach is to facilitate a “good-contact” between In-MOF and GO, which is crucial in enhancing photocatalytic performances of heterostructure. Instead, when using an ex-situ approach to fabricate In-MOF/GO heterostructure from the mixed suspension of GO and pre-synthesized In-MOF (referred to as In-MOF/GO-ex-situ; Supplementary Fig. 4), its activity was very limited, with no notable improvement compared to individual In-MOF or GO (Fig. 2b). Moreover, the duration of in situ growth remarkably affects the reaction performance29, as evident from Supplementary Fig. 5. Among the heterostructures with different in situ growth durations, In-MOF/GO-4h exhibits the highest photocatalytic performance, highlighting the importance of kinetically controlled growth of In-MOF on GO.
To confirm the source of CO generation, control experiments were conducted without light, photocatalyst, or CO2; none of these experiments generated CO (Supplementary Fig. 6). In addition, using 13CO2 instead of 12CO2, the peak at m/z = 29 (13CO) in the MS spectrum confirms that the source of C in CO originates from CO2 (Fig. 2c). Further, the stability of In-MOF/GO-4h was tested; even after five cycles of aerobic CO2 photoreduction, the catalyst still maintains 95% efficiency (Fig. 2d). Characterization of In-MOF/GO-4h after five cycles shows almost undetectable variations in the morphological, crystallographic, or chemical structure and no detachment or aggregation of the In-MOF nanosheets (Supplementary Figs. 7, 8, and 9). Next, the intrinsic relationship between O2 concentration and CO2RR rate was explored. The results reveal that, compared to an oxygen-free environment, the decline in the efficiency of aerobic CO2RR efficiency is very limited. Notably, even when the oxygen concentration reaches 20%, In-MOF/GO still exhibits remarkable efficiency and the CO generation rate decreased by only 5% compared to that in an oxygen-free environment. This finding demonstrates the excellent tolerance and efficiency of In-MOF/GO in performing the CO2RR under aerobic conditions, maintaining a high catalytic activity despite the interference from elevated levels of oxygen.
Furthermore, to monitor the products of oxidative half-reactions in the In-MOF/GO-4h loaded artificial leaves, we measured the generated O2 during photoreaction in an oxygen-free atmosphere containing 10% CO2 and 90% Ar. Along with the generation of CO from CO2 reduction, O2 derived from water oxidation was also observed. The generation of products follows a linear growth that persists for 12 h (Fig. 2f). The observed ratio between the generation rates of CO (762.5 µmolཥg− 1ཥh− 1) and O2 (321.8 µmolཥg− 1ཥh− 1) is higher than the corresponding stoichiometric ratio of 2, suggesting the existence of another product from the oxidative half-reactions. Therefore, we next measured the possible liquid products of the photocatalytic reactions. No liquid products, such as formic acid, are generated by the CO2RR half-reaction; however, H2O2 is generated at a generation rate of 212.5 µmolཥg− 1ཥh− 1. Notably, the amount of generated H2O2 is not affected by the O2 content in the atmosphere. Only a slight reduction of 5% in H2O2 generation rate is observed when the O2 ratio in the atmosphere increases from 0–20% (Supplementary Fig. 10). Therefore, H2O2 is mostly likely generated by water oxidation. This further explains the discrepancy between the observed ratios of generation rates and stoichiometric values between CO and O2 and when comparing to the literature reports, this floating system exhibits superior activity in the H2O2 generation originated from water oxidation30–32.
Owing to the generation of H2O2, a widely used reagent in aqueous pollutant degradation and sterilization, on the floatable artificial leaves, we considered coupling aerobic CO2 reduction with water purification for real environmental applications. We collected lake water from Beijing Olympic Park and used it with our floatable device in an atmosphere containing 10% CO2, 20% O2 and 70% Ar. Supplementary Fig. 11 shows the photocatalytic reactor setup used for purification of water. In both deionized water and lake water, the floatable device exhibits identical performance in the aerobic photocatalytic CO2 reduction to CO, indicating that the presence of organic or inorganic substances in lake water does not interfere with CO2 reduction activity (Fig. 2g, upper part). Next, we monitored the variations in Chemical Oxygen Demand (COD) of lake water during the photocatalytic reaction. The COD values (Fig. 2g, lower part) decrease linearly with illumination time, suggesting the decomposition of organic contaminants by the in situ generated H2O2 or photogenerated holes. After 6 h, the COD of the lake water falls below 15 mg/L, meeting class I water standard of China (Supplementary Fig. 12). Hence, the design of the floatable artificial leaves provides a facile reaction system suitable for use in open water environments.
Investigation of preferential CO adsorption
Next, we investigated the origin of the aerobic photocatalytic CO2 reduction by In-MOF/GO. First, we investigated electron transfer between the two moieties of In-MOF/GO. Density functional theory-based calculations performed using the In-MOF/GO heterostructure model show that electron densities on the GO and In-MOF moieties decrease and increase, respectively, indicating electron transfer from GO to In-MOF during the formation of the heterostructure (Supplementary Fig. 13). This theoretical result corroborates well with the X-ray photo-electron spectra (XPS) results. In the In-MOF/GO composite material, a shift in the C 1s peak of GO toward higher binding energies occurs simultaneously with the counter-directional shift of the In 3d peak of In-MOF. This phenomenon indicates electron transfer from GO to In-MOF within the heterostructure (Supplementary Fig. 14).
In the theoretical model of In-MOF/GO, after placing a CO2 molecule and subsequent structural optimization, the analysis of electron density overlap between the CO2 molecule and axial hydroxyl on the In node in In-MOF reveals the capability of the surface hydroxyl to capture molecular CO2. After the attachment of CO2 to the hydroxyl group of the In-node in the In-MOF/GO heterostructure, CO2 receives partial electrons from both GO (0.40 e−) and In-MOF (0.05 e−) moieties (Fig. 3a). In this case, the total electrons transferred (0.45 e−) to CO2 are significantly higher than those when CO2 is attached to a self-supported In-MOF (0.13 e−), as depicted in Fig. 3b. Thus, the adsorption energy of CO2 on an In-MOF/GO heterostructure sharply decreases to -0.57 eV, indicating a more stable CO2 capture compared to the adsorption energy of CO2 on a self-supported In-MOF (-0.11 eV), as shown in Fig. 3c. Investigations on the interactions between O2 and In-MOF/GO reveal that the surface sites of In-MOF/GO, including the surface hydroxyl groups, are almost inert to O2 adsorption (endothermic by approximately 0.09 eV). In conclusion, the heterostructure of In-MOF/GO significantly enhances the capture of CO2 over O2, which is crucial for aerobic photocatalytic CO2 reduction.
The interaction between CO2 and the surface hydroxyl groups of the In-node of In-MOF was further analyzed using in situ Fourier Transform infrared spectroscopy (FT-IR) (Supplementary Fig. 15). The FT-IR results show that when CO2 is introduced, the intensity of the IR peak located at 3612 cm− 1, attributed to the hydroxyl on the In node33, gradually decreases. Simultaneously, a new band at 2347 cm− 1 emerges and gradually becomes prominent. This band, which is different from the doublet gaseous CO2 band, has a singlet feature and frequency close to those of dissolved CO2 in an aqueous solution (solvated CO2 by forming hydrogen bonds with the water solvent)34. Therefore, this band can be assigned to CO2 interacting with surface hydroxyl groups via hydrogen bonding. The FT-IR results further confirm the interaction between the hydroxyl groups on the In-node and CO2, facilitating CO2 capture on In-MOF/GO. In conclusion, the surface hydroxyl groups on In-MOF/GO facilitate the selective capture and enrichment of CO2.
The adsorption–desorption curves of CO2 and O2 on In-MOF/GO at 1 atm show adsorption capacities of 18.21 cm3/g and 1.62 cm3/g for CO2 and O2, respectively (Fig. 3d). Considering the composition of the gaseous atmosphere used in our aerobic photocatalytic CO2 reduction, we compared the results obtained at 0.1 and 0.2 atm for CO2 and O2, respectively. Analysis of the initial slopes of the adsorption isotherms at these specified pressures reveals an approximate selectivity ratio of 30 for CO2 versus O2 adsorption on In-MOF/GO. This finding strongly supports the superior adsorption capacity of In-MOF/GO for CO2 compared to O2.
Photocatalytic reaction mechanism
To elucidate the mechanism of CO2 reduction on In-MOF/GO, we collected in situ FT-IR spectra during the photocatalytic reactions. The reaction was first conducted under oxygen-free conditions in a D2O-saturated gas atmosphere containing 10% CO2 and 90% Ar. The strategic use of D2O instead of H2O precludes the masking effect of the intense H2O bending vibration band (~ 1630 cm− 1), enabling a clear identification of the characteristic bands from the MOF structure, such as the carboxylate/carboxylic acid moiety. Additionally, a parallel 13C-isotope labeling experiment was conducted using 13CO2 instead of 12CO2 to identify the origin of the IR band, from CO2-related intermediates or structural variations in the MOF structure. Upon illumination, a gradual depletion of intensity at 1206 cm− 1, attributed to the bending vibration of D2O, indicates the oxidative consumption of water (Fig. 4b). Based on the presence of 13C-isotope shift, the newly emerged bands can be classified into two groups. The bands at 1631 and 1376 cm− 1 exhibit their 13C-counterparts at 1599 and 1344 cm− 1, respectively (Fig. 4c). These bands can be assigned to *COOH35–37, a vital intermediate in the reduction of CO2 to CO. Notably, the band at 1631 cm− 1 observed under 12CO2 exhibits a right shoulder at approximately 1658 cm− 1. Under 13CO2, the band at 1631 cm− 1 shifts to 1599 cm− 1, appearing as a negative peak. Based on the standard IR spectrum of In-MOF, the band at 1658 cm− 1 (Fig. 4a) can be attributed to the OCO vibration in the carboxylate-coordinated In-node (COO-In)38. The depletion of the carboxylate-related band at 1658 cm− 1 is accompanied by the growth of the band at 1760 cm− 1. This is a characteristic of the OCO vibration in carboxylic acid and is attributed to the protonated form of carboxylate. Considering the theoretical prediction and XPS results that in the hybrid structure of In-MOF/GO, the electron transfer from GO to In-MOF moiety, it can be speculated that upon illumination, the In-node in In-MOF trapped the photogenerated electrons and being reduced. The reduced valance state of In results in reduced coordination numbers, causing the partial dissociation of the coordinated carboxylates (IR band at 1658 cm− 1), which were further by protons released from water oxidation to form carboxylic acid (IR band at 1760 cm− 1). The FT-IR results confirm that the In nodes in the In-MOF are the electron-enriched sites that are formed during photocatalytic reactions and are available for subsequent CO2 reduction. Notably, the structural variations in the In-nodes are reversible. After cessation of illumination, the intensities at 1658 and 1760 cm− 1 can be observed to recover, indicating restoration of the initial structure of the In-nodes (black spectrum in Fig. 4b).
The in situ FTIR-IR results also confirm that this tandem process was not affected by the presence or absence of O2. When the photocatalytic CO2 reduction is conducted under aerobic conditions (atmosphere: 20% O2, 10% CO2 and 70% Ar, saturated with D2O), the intensity ratio between 1658 and 1760 cm− 1 bands can be observed to vary (Fig. 4d). The corresponding intensity depletion of the D2O-related band and appearance of CO2RR intermediates-related bands can also be clearly observed (Fig. 4d). All emerged bands display an analogous behavior similar to those of the corresponding bands observed under anaerobic conditions (Fig. 4b). The observed similarities in the behavior of the bands confirm that the catalyst under study retains its efficiency in performing photocatalytic reactions, even under aerobic conditions. Based on in situ FT-IR spectroscopy, the mechanism of photocatalytic CO2 reduction process on In-MOF/GO is depicted in Fig. 4e. It can be concluded that the In-node in the In-MOF/GO heterostructure plays an important role in both CO2 capture and reduction. CO2 is initially captured by the hydroxyl groups of the In-nodes. The captured CO2 is then reduced to *COOH at the photo-reduced In-nodes, leading to the generation of CO, and the reduced In- nodes recovers for the next catalytic cycle.
Fabrication of a heterostructure between GO and In-MOF is also essential for accelerating CO2 reduction. The theoretical calculations (Fig. 3a) demonstrate a more significant electron transfer to the CO2 adsorbed on an In-MOF/GO heterostructure than on a self-supported In-MOF, activating chemically inert CO2. Further simulation of CO2 reduction to *COOH via coupled electron/proton transfer shows that the energy barrier for this step on an In-MOF/GO heterostructure is 3.06 eV, which is 0.56 eV lower than that on a self-supported In-MOF (Supplementary Fig. 16). This substantial difference in energy barriers indicates that CO2 reduction proceeds more easily on the In-MOF/GO heterostructure, further highlighting the remarkable efficacy of in situ fabricated In-MOF/GO heterostructures in photocatalytic applications.
{kind=link}