Accumulating glacial ice contains a time-sequenced record of atmospheric constituents from a time in the past, including viable and non-viable cells that remain preserved in the ice. Although small subunit ribosomal RNA amplicon sequencing has been a useful tool to characterize glacial ice assemblages [1-3], the limited phylogenetic and genetic resolution of this approach poses significant limitations for functional insights. Accessing the metagenome of glacial “fossils” can provide a uniquely powerful historical record of microbial genetics from varying climate conditions (e.g., [4]) and predate human activities that have transformed landscapes and land cover. In this study, we sought to validate techniques for recovery and sequencing of ancient genomes preserved in samples from an alpine glacier ice core that contained low biomass (~105-107 cells L-1; Fig. 1B).
The Wind River Range (Wyoming, USA) is one of few locations in the continental US where a detailed paleoecological glacial record is available [5-8]. Two ice cores recovered from the Upper Fremont Glacier (UFG) are archived by the National Science Foundation Ice Core Facility. The samples analyzed in this study were obtained from the ice core drilled in 1998 (43.1294444, -109.6163889) designated FRE98-4. Ice cores from the UFG contain an ~250-year record of climate and anthropogenic pollution for the contiguous United States that extends to the middle of the 18th century (1746 to 1998 CE for FRE98-4) [5-8]. This coincides with the expansion of cultivated land area in the American West, as well as notable climatic events (i.e., termination of the Little Ice Age in 1870 CE and Dust Bowl drought of the 1930s) and increases in local air temperature (Fig. 1A).
Analysis of ice samples from 28 depths using previously described methods [9] revealed a two orders of magnitude microbial cell concentration range (Fig. 1B). There is decanal to century scale variation in annual layer thickness in the FRE98-4 core. At our sampled depth resolution (17.5 to 90 cm), each sample represents a portion to annual year of deposition. The 70.9 mbs (meters below surface; 1940 CE) sample contained the highest observed cell concentration while lower concentrations were generally observed samples corresponding to the Little Ice Age. Ice core depths shallower than 117 mbs contained a record of biological aerosols from regional ecosystems during the early to late 20th century. In samples originating from 1904 to 1966 CE (n=16), cell concentration is positively and significantly correlated to sample age (Spearman’s rank order correlation, r(14) = 0.603, p < 0.05). Although previous studies have shown positive correlations between cell and dust or black carbon concentrations in glacial ice [10-13], the variation in dust and black carbon data from FRE98-4 are dissimilar to the trend of increasing cell concentrations after 1904 CE (Fig. 1). Based on δ18O-H2O data (Fig. 1A), local air temperature since the termination of the Little Ice Age (1870 CE) to the early 1990s has increased by ~5oC [5]. This period of warming coincided with large scale changes in land use and cover that accompanied the intensification of agriculture in the western United States (Fig. 1A).
To examine variation in the microbial assemblages preserved with depth/age in the ice, we extracted DNA and sequenced metagenomes from six relatively large ice samples (650-920 g cleaned weight; Table S1). Based on top depths from 26.87-153.01 mbs the samples span 170 years (1961-1790 CE; Fig. 1). Aseptic sampling and cleaning of ice cores samples followed previously described methods and a mock sample of frozen sterile ultrapure water was processed as a control [9]. Melt water was filtered with Centricon-70 30 Kda devices by repeated 10-15 minute centrifugation cycles at 3,500 RCF using a swinging bucket rotor. We used three techniques to recover DNA. First, 10% of the recovered volumes were used directly as environmental DNA (eDNA). The remaining 90% volume was split evenly and processed using either the MP FastPrep FastDNA SPIN for Soil kit (FP) according to the manufacturer’s recommendations, or the Promega Wizard HMW DNA (HMW) for Gram negative bacteria with 80°C lysis treatment. Although, instead of ethanol precipitation, clarified lysate was filtered through a pre-washed Amicon Ultra-2 30 kDa filter and washed twice with TE buffer (pH 8.0) prior to retentate recovery. Samples were then amplified with the Takara PicoPLEX Single Cell WGA Kit v3 according to the manufacturer’s recommendations. Pure PicoPLEX reagent water was amplified as an additional negative control (PCRneg). DNA amplification yielded nine samples with fragment sizes >400 bp and DNA concentrations >10 ng/μL (Table S1). These were sequenced by SeqCenter using the on-bead Tagmentation Illumina DNA prep with the small shotgun metagenomic sequencing pipeline, targeting 6.5M 150-bp paired-end reads/sample. As expected, the control samples had low DNA concentrations (Table S1) and were sequenced using low-DNA input of the Nextera Flex for Enrichment library prep kit followed by MiSeq Illumina sequencing with a Nano flow cell, targeting 2M paired-end 250-bp reads/sample at the Georgia Genomics and Bioinformatics Core.
For metagenomics analysis, adapter and quality trimming was performed with Trimmomatic [14], with a sliding window trim of 4 bp, minimum quality 30, leading and trailing quality cutoffs of 3, and a minimum length of 36 bp. Alpha and beta diversity were determined by rarefying samples to 140,000 reads, trimming to a maximum of 150 bp/read, and calculated with Krakentools [15]. Taxonomic classifications were performed with Kraken2 as well as Bracken at the genus level [16, 17]. All of the ice core samples show higher diversity than the negative controls (Fig. 2A), with a trend of lower diversity in the 1900-1948 CE samples. The controls clearly separated from the glacial samples based on Bray-Curtis ordination (Fig. 2B). The sample depths 116, 108, 69, and 51 mbs covering a ~50 year period (1900-1948 CE) had similar beta-diversity ordination although cell concentrations varied substantially over this time frame. The method of DNA extraction did not have an obvious effect on the recovered microbial community, with highly similar compositions inferred by each of the methods (Fig. 2B). The eDNA method produced the most samples suitable for sequencing (4 of 9) and required the least processing and thus may be a preferable choice for future metagenomic sequencing of glacial ice.
Taxonomic profiles were calculated with Kaiju [18]. The most common and abundant taxa were bacteria in the Actinomycetota, especially Dietzia and Nesterenkonia (Fig. 2C). Over half of the most abundant genera were enriched in the oldest sample (153 mbs; 1790 CE). These include Dokdonella, Ginsengibacter, Hanamia, Nitrosospira, and Rhodanobacter, which on average, are present at >10-fold the abundance as in the other samples, and Gemmatirosa and Polaromonas, which were an average of 3 to 5-fold more abundant. Many species of Polaromonas are psychrophilic [19] and their presence could be consistent with the cooler Little Ice Age climate of 1790s. In addition, some ammonia oxidizing and denitrifying members of the Nitrosospira [20] and Rhodanobacter [21] are also known to be cold-adapted.
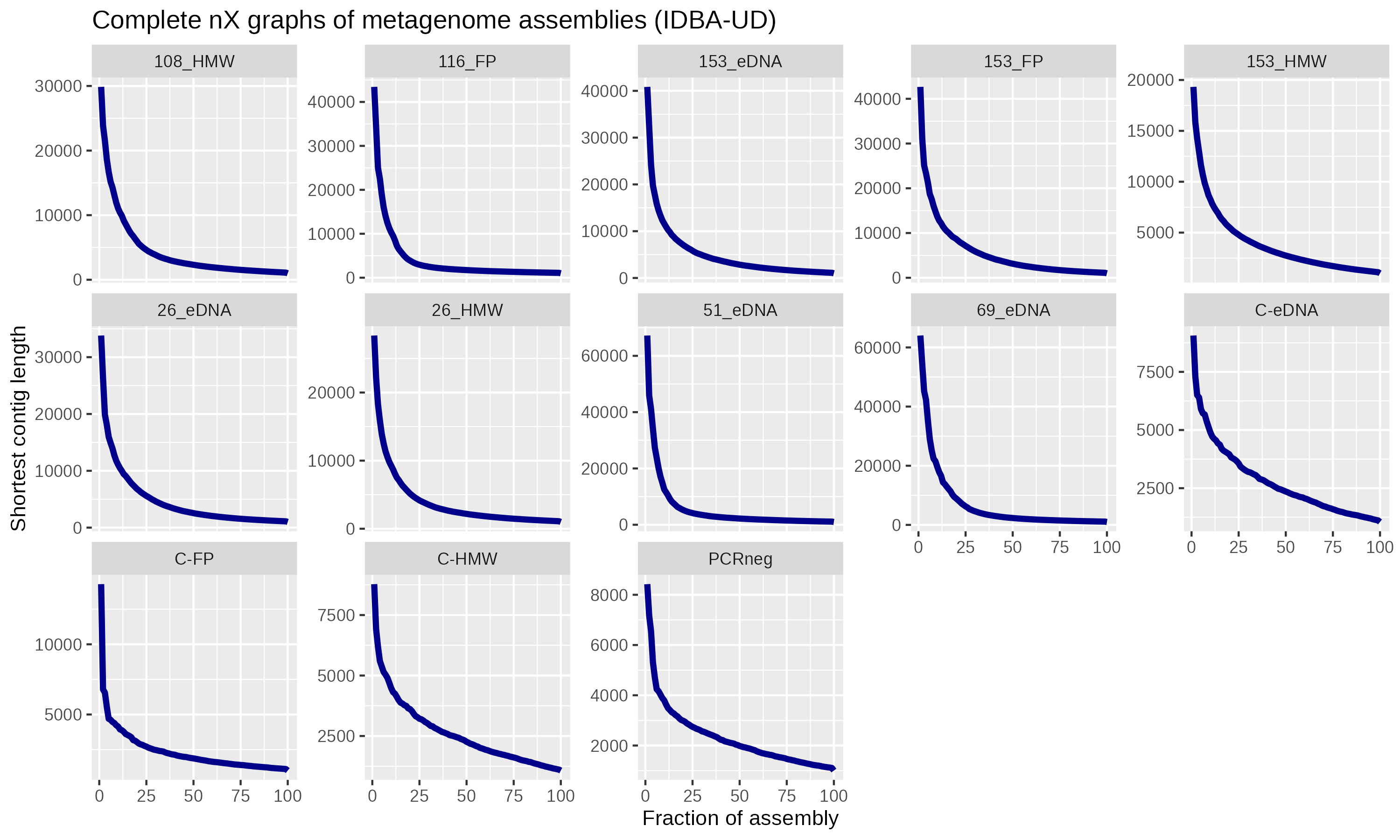
Metagenome assemblies performed with IDBA-UD [22] were highly fragmented (Fig. S1). To filter out contaminating sequences in the assembled metagenomes, we aligned the reads from each to the 4 control samples using bwa [23]; any contigs with >10% coverage or >1x average depth from control assembles were removed. Functionality was determined with DRAM [24]. The most pronounced pattern was the enrichment of nitrogen-cycling genes in the 153 mbs samples (Fig. 2D) and may be related to the abundance of Nitrosospira and Rhodanobacter (Fig. 2A). Temporal patterns in antibiotic resistance genes were assessed by matching contigs against the Comprehensive Antibiotic Resistance Database [25]. We identified the efflux pump gene adeF, dihydrofolate reductase dfrB10, and several vanY D,D-carboxypeptidase genes (Fig. S2). However,asthese genes can each participate in other cellular functions, their significance to antibiotic resistance is unclear.
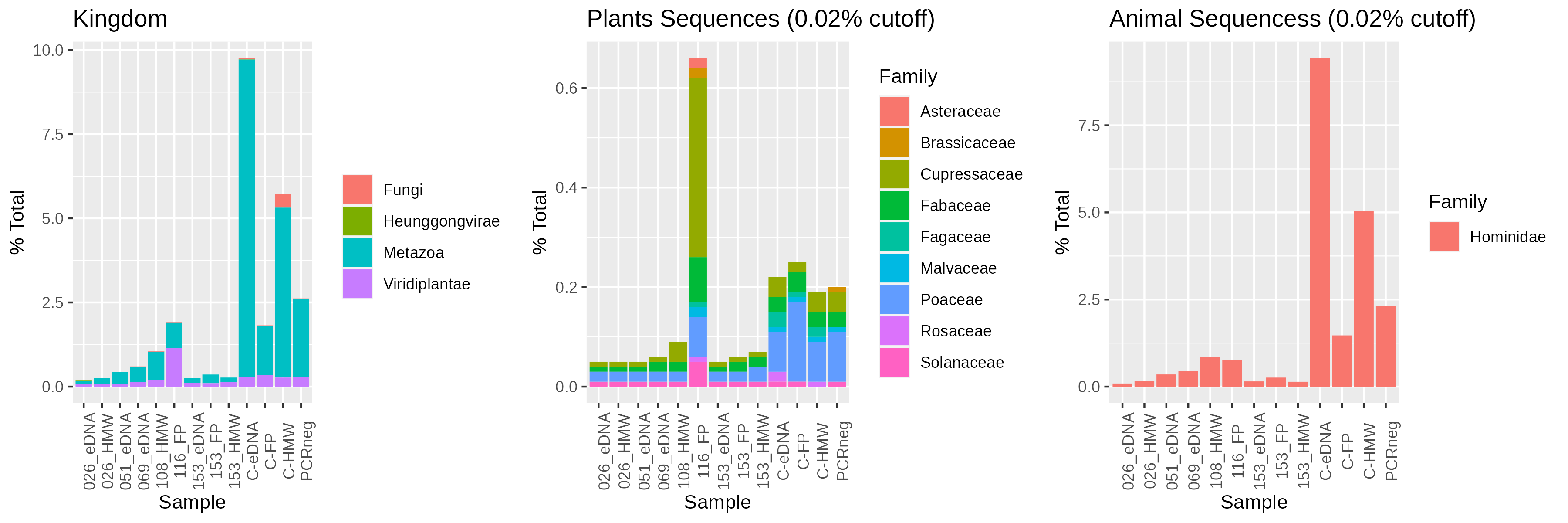
We identified an excess of plant sequences in the 116 mbs (1900 CE) sample (Fig. S3). While Kraken2 flagged a significant portion, (0.36% of total and 31% of plant sequences), as Cryptomeria japonica (Japanese Cedar), manual BLAST indicated sequences as similar to Pinus sp. (data not shown), which are widely distributed in the western United States [26]. This spike in plant signal coincides with low microbial cells count (Fig. 1B), moderate alpha diversity (Fig. 2), and the period when temperatures increased into the 21st century [5] (Fig. 1A).
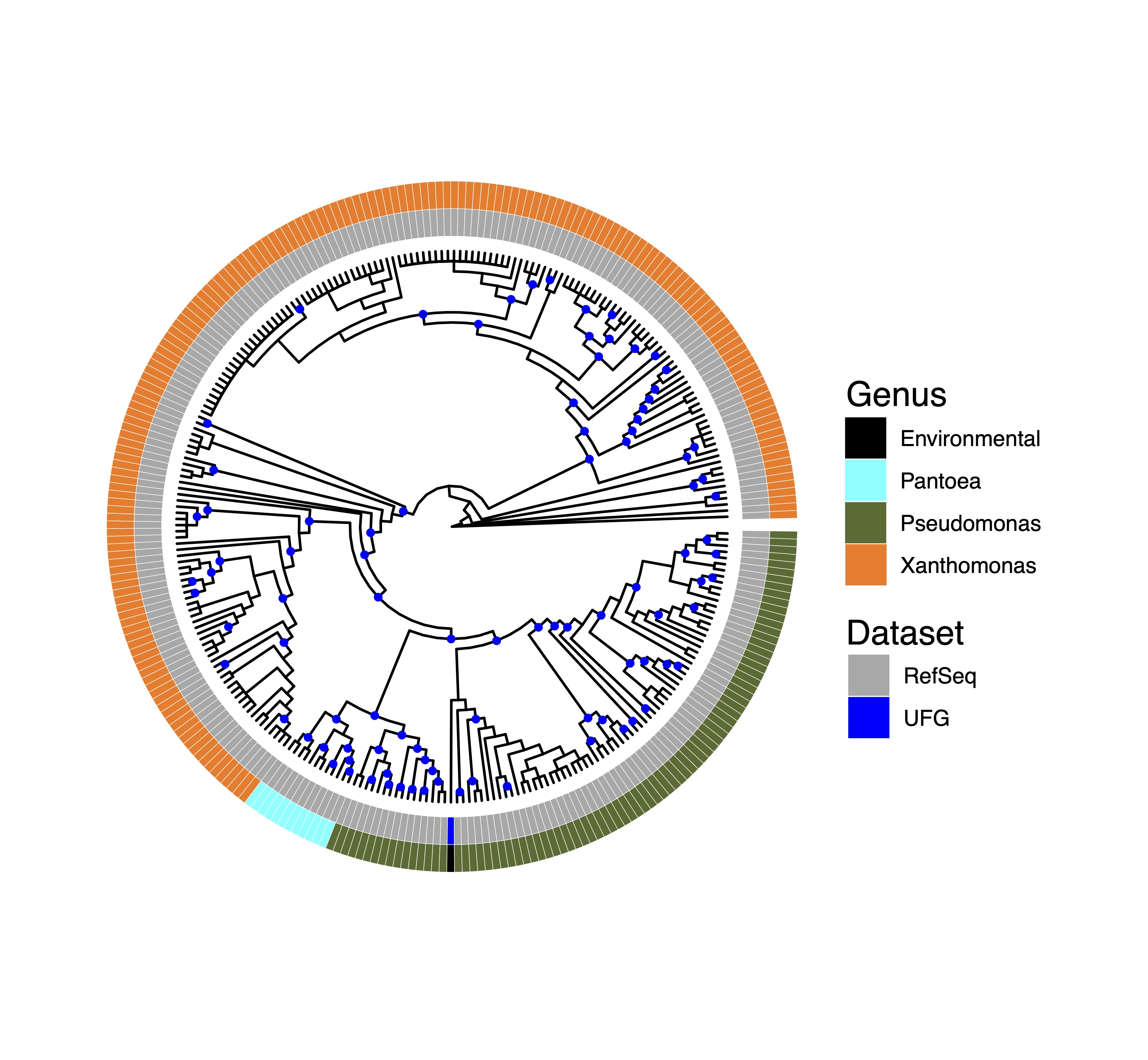
We hypothesized that microbes immured in glacial ice would be enriched for ice nucleating activity (INA) genes due to their known role in bioprecipitation [27]. Metagenomic contigs were annotated using Prokka and a custom INA Hidden Markov Model (HMM) generated with hmmer [28] trained on twelve INA proteins aligned with muscle [29] was used to identify INA genes. A single INA gene was identified from the 153 eDNA sample that phylogenetically clusters with inaZ from Pseudomonas (Fig. S4).
In conclusion, we present methods to extract, amplify, assemble and analyze metagenomic DNA from cells and eDNA preserved in glacial ice cores. Our results indicate that direct amplification of environmental DNA (the eDNA method) was reliable and the simplest approach for recovering metagenomics sequences. Further improvements to this method should focus on increasing the sample sequencing depths and contiguity.
{kind=link}
{kind=link}
{kind=link}
{kind=link}