Materials
Lipoic acid, 1,1'-carbonyldiimidazole (CDI), L-Arginine methyl ester dihydrochloride, N, N-diisopropylethylamine, Triethanolamine (TEOA), and 2-Iodoacetamide were obtained from Aladdin Biochemical Reagents Co., Ltd. (Shang Hai, China). Dimethylformamide (DMF), Dichloromethane (DCM), and Ethyl ether (Et2O) Reagent Co., Ltd. All chemicals were of analytical grade and used without any further purification. The ReverTra Ace qPCR RT Master Mix with gDNA remover kit and SYBR qPCR Mix were purchased from TOYOBO (Japan), all Restriction enzymes, T4 ligase, Grace’s insect medium, Fetal bovine serum (FBS), and penicillin − streptomycin were purchased from Invitrogen (Carlsbad, CA, USA), All primers were synthesized at Qingke Biotechnology Co., Ltd (Bei Jing, China).
Insects and cell culture
The Sf9 cells, originating from ovarian tissue of female fall armyworm pupae, were cultured in Grace’s insect medium with 10% fetal bovine serum (FBS) and 1% penicillin − streptomycin at 27 ± 1°C.
FAW eggs were obtained from Shanghai Tahoe Chemical Co., Ltd (Shang Hai, China). They were maintained on corn seedlings in a climate-controlled incubator at 27 ± 1°C with a photoperiod of 16:8 (light: dark). Larvae were fed an artificial diet.
Construction of plasmid vectors for expression of dsRNA and extraction of dsRNA
The protocol of construction of plasmid vectors of dsRNA and the isolation of RNA from Escherichia coli was adapted in the published research(Murphy et al. 2016; Posiri et al. 2013). In brief, the total RNA was extracted from four third-instar nymphs of FAW by using the RNAiso Plus solution. Further, cDNA was synthesized by RT-PCR using the ReverTra Ace qPCR RT Master Mix with gDNA remover kit. Two Fragments, VG-L and VG-R for generating dsRNA, were amplified with primers from cDNA with primers shown in the Table S1, respectively, using the following program: 30 s at 94°C, 30 s at 55°C, and 10 s at 72°C for 30 cycles. The VG-L contained a stem–loop structure was digested with Bam HI–Eco RI and digested with Eco RI–Xho I to generate the complementary VG-R. Products were purified by Omega Gel Extraction Kit and were cloned into the pET-28a digested with Bam HI–Xho I together. Then, pET28a was transformed into BL21(DE3) RNase III- which previously knocked out the rnc gene encoding endoribonuclease. After cells were cultured at 37°C overnight, a single colony was verified to be correct and cultured in 5 ml of LB liquid medium with kanamycin (50 mg/mL) overnight and diluted into the 50 ml shake flask. 1 mM IPTG was added and cultured for 4–5 h at 37°C when the OD600 value reached 0.6. The cell culture medium was treated at 80°C for 20 minutes, then centrifuged at 12,000 rpm for 3 minutes. 75% absolute ethanol mixed with phosphate-buffered saline (PBS) was added and centrifuged at 8000 rpm for 4 minutes. 250 µL of 150 mM NaCl solution was added to resuspend the cells, and the cell culture was centrifuged at 12 000 rpm for 2 min to obtain the coarsely extracted dsRNA. RNase-free DNase (5 µL) and RNase A solution (2 µL) were added to 250 µL of the dsRNA solution for 30 min to remove from DNA and single-stranded RNA. The solution was purified with magnetic beads according to the manufacturer’s programs and then quantified using a NanoDrop 2000 spectrophotometer.
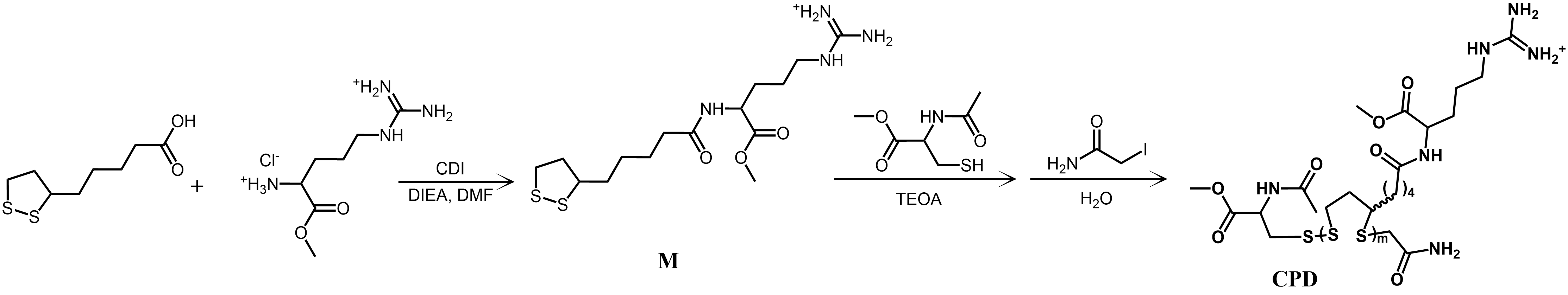
Synthesis of M
Following a slightly adapted procedure(Bang et al. 2013), a solution of lipoic acid (2.06 g, 10 mmol) in anhydrous DMF (20 ml), CDI (1.62 g, 10 mmol) was added, and the mixture was stirred at 25℃ under N2 atmosphere for 2 h. A solution of L-arginine methyl ester dihydrochloride (1.31 g, 5.0 mmol) and N, N-Diisopropylethylamine (0.87 mL, 5.0 mmol) in anhydrous DMF (20 ml) was added, and the reaction mixture was continued to stir at 25℃ under N2 atmosphere for 3.5 h. Afterward, the reaction mixture was added dropwise into Et2O (100 ml). The mixture was centrifuged (3 min, 3000 rpm), and the supernatant was discarded. The remaining yellow oil is washed three times with a DCM/Et2O mixture (1:2, 5×10 ml). After drying the residue in vacuo, a pale yellow solid M (346 mg, Cl salt, 84%) was obtained. 1H NMR (400 MHz, Deuterium Oxide) δ 4.30 (dd, J = 9.2, 5.0 Hz, 1H), 3.63 (m, 4H), 3.10 (m, 2.9 Hz, 4H), 2.54–2.14 (m, 4H), 1.84 (dd, J = 14.8, 6.9 Hz, 2H), 1.72–1.63 (m, 2H), 1.58–1.51 (m, 4H), 1.38–1.27 (m, 2H).
Synthesis of cell-penetrating disulfide polymer (CPD)
M and initiator, the N-acetyl-L-cysteine methyl ester, were dissolved into the TEOA buffer solution (1.0 M, pH = 7), respectively. For polymerization, the M (100 mM) and initiator 4 (0.2 mM) are stirred vigorously at 25°C under an N2 atmosphere in 20 ml TEOA buffer. After a fixed 30 min, the mixture was quenched with a terminator solution (40 mL, 250 mM) in water and purified by dialysis in water for 3 days, and then, the CPD was obtained. The structure of CPD was validated by 1H NMR (400 MHz, D2O), and the relative molecular weight was analyzed by gel permeation chromatography (GPC).
Gel retardation assay
The ability of CPD to condense dsRNA was evaluated through gel electrophoresis. The complexes were incubated at mass ratios of 1:4, 1:2, 1:1, 2:1, 4:1, 6:1, and 8:1 by loading 0.25, 0.5, 1, 2, 4, 6, and 8 µg of CPD with 1 µg dsRNA for 30 min at room temperature. The mixture was estimated with 1.0% agarose gels made from 1×TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH = 8). The gel electrophoresis was carried out at 150 v for 20 min.
Particle size and zeta potential analysis.
The CPD was dissolved in water to form nanoparticles with the dsRNA. The particle size and zeta potential of the CPD and CPD/RNA complexes were analyzed in triplicate at 25°C with a Zetasizer Nano ZS. The appearance of the CPD/RNA complexes was observed by transmission electron microscopy (TEM).
dsRNA Protection Performance.
The protection of dsRNA from nuclease degradation was evaluated using RNA enzymes. Briefly, naked dsRNA solution (1600 ng) and CPD/dsRNA were incubated with 25 U of nucleic acid allozyme at 37°C for different periods (0, 2h) in triplicates. Following incubation, 100 mM NaBH4 was added instantly to released dsRNA, and 0 h of dsRNA was used as the control. The different dsRNA samples were extracted and subjected to 1% agarose gel containing Gel Blue stain, followed by quantitative analysis using Quantity One software.
Cell viability assay
The cells were transfected with different concentrations of nanoparticles to characterize cell viability, which was evaluated using the CCK-8 assay kit. Briefly, Sf9 cells are seeded into 96-well plates at a density of 5 × 104 cells per well. After seeding for 2 h, the CPD and CPD/dsRNA complexes were transfected in 100 µl culture medium. The concentration of CPD and CPD/dsRNA complexes varied from 0 to 64 µg/ml, and the quantity of dsRNA was fixed at 100 ng per well. After incubation for 24 and 48 h, respectively, the medium was removed, and 100 µL fresh medium containing 10 µL of CCK8 solution was added into each well with PBS-washed medium for 1.5 h. The absorption of each well was measured at 450 nm with a microplate reader to calculate the OD values. The cell viability was calculated by the formula:
\(\:Cell\:viability=\frac{\left(A-{A}_{0}\right)}{({A}_{1}-{A}_{0})}\times\:100\) %
where A represents the absorbance of the cells treated with various complexes,
A1 represents the absorbance of medium blanks, and A0 is the absorbance of the cells without any treatment.
Plasmid construction and mRNA in vitro transcription
The plasmid PIB-V5-His (pDNA)and mRNA encoding Green Fluorescent Protein (GFP) were constructed to test the cell transfection efficiency on the Sf9 cell line. Cells were seeded in 24-well plates at a concentration of 1.5×105 cells/well in 0.25 mL of SF 900 II medium and incubated overnight at 27°C. CPD was incubated with a pDNA and mRNA (500 ng) solution at a mass ratio of 6:1 for 30 min at room temperature to prepare the CPD/pDNA and CPD/mRNA complexes. Then, the complexes were added to the cell medium. The PEI/pDNA solution, according to the manufacturer’s protocol, was added. pDNA alone and CPD were set as the controls. After 48 h of transfection, the green fluorescence was used to reflect the delivery of CPD into insect cells by fluorescence microscopy.
Internalization of CPD-conjugated CypHer-5E-labeled dsRNA
In the current work, CypHer5E-labeled dsRNA was labeled in red, Lyso-Tracker™ was employed to stain lysosomes in green, and the Hoechst33258 was used to label the nuclear in blue to explore the cellular uptake and lysosomal escape of CPD. 4×105 Sf9 cells were exposed to the proper amounts of CypHer5E-labeled dsRNA-CPD in 1 ml of serum-free culture medium at 2 h,4 h, and 6 h, respectively, and the naked CypHer-5E-labeled dsRNA was used as a control. Then, the cells were stained by Lyso-Tracker™ and Hoechst 33258, respectively. The images were scanned by Nikon A1 confocal laser scanning microscopy.
Bioassays
The larvae of FAW were fed on an artificial diet for the duration of the assay; the second instars with the same body size were placed in the sterile Petri dish with a diameter of 5 cm for the later RNAi experiment. 10 instars were placed in each plate, and the dsRNA at a concentration of 1000 ng/µL and CPD were mixed to make the artificial diet, and then fully mixed with artificial food of about 1 cm3 and then evenly added to the Petri dish. The ones treated with CPD/dsGFP and CPD were used as the control group, and each experimental group had 5 replicates. There are 50 nymphs in total. The nymphs were put into the incubator at 26 ± 1°C and the photoperiod of 14:10 h. The food was changed every 2 days and was stopped after 6 days, and the normal food was changed back to continue feeding. Changes in body weight and body length in each treatment group are recorded on the sixth day, and insect mortality is recorded on the tenth day.
Interference Efficiency analyzed by qPCR
Total RNA was extracted from larvae using RNAiso Plus reagent, and cDNA was synthesized with one microgram of total RNA using a reverse transcription kit. The relative mRNA levels were performed by the 2−ΔΔCt method(Livak et al. 2001), and the RpL10 was the reference gene. The primers are shown in Table S1. qPCR was performed with SYBR Green Supermix, and the reaction system contained 10 µL of SYBR qPCR Mix, 1 µL cDNA, 0.4 µl each of 10 µM forward and reverse primers and 8.2 µL of RNase-Free water in 20 µL final volume. The reaction conditions were followed by 95°C for 30 s, 40 cycles of 95°C for 5 s, and 60°C for 30 s settings.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism version 9 software (GraphPad Software Inc.). All statistics were shown as the mean ± standard deviation (SD). The Student’s t-test for two samples and one-way ANOVA with a Tukey’s post-hoc test for more than two groups were applied. P < 0.05 is considered statistically significant (*P < 0.05, **P < 0.01, and ***P < 0.001).
{kind=link}