Data and code availability
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information, except for the raw data for scRNA-seq reported in this publication can be accessed under the Gene Expression Omnibus (GSE182159) and the Genome Sequence Archive (https://ngdc.cncb.ac.cn, accession number HRA001730) on request. This paper analyses existing data. These accession numbers for the datasets are listed in the key resources table.
Any additional information is available upon request.
Animal models
All the animal experimental procedures were approved by the Animal Care and Use Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, with project license numbers of SH9H-2021-A79-1 (bone marrow organoid associated animal experiments) and SH9H-2023-A778-1 (islet organoid associated animal experiments). Mice were housed in the specific pathogen-free facility at Shanghai Ninth People’s Hospital under a 12-hour light/12-hour dark cycle (06:00 to 18:00) at room temperature of 22 ± 1 °C, with ad libitum access to food and water.
For subcutaneous implantation of RMF-derived chondroid pellets, NOD/LtJ-PrkdcscidIl2rgem1/Shjh female mice aged 4 weeks were used. For secondary transplantation of hCD34+ cells, NOD/LtJ-PrkdcscidIl2rgem1/Shjh female mice aged 8-10 weeks were used. For kidney subscapular implantation of RMF-derived islet organoids, NOD.Cg-Prkdcscid/Shjh with aged 8-10 weeks were used. Mice of the same sex were randomly assigned to experimental groups. Both strains of mice were purchased from Shanghai Jihui Laboratory Animal Care Co., Ltd.
Human subjects
Human adult adipose tissue samples were collected from health individuals who underwent liposuction surgery (median age: 31 years, range 20-50). Donor information was shown in Supplementary Table 1. The collection and use of human adult adipose tissue samples were approval by the Institute of Ethics Committee Review Board in Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (SH9H-2023-TK189-1), in accordance with the principles outlined in the declaration of Helsinki. Informed consent was obtained from the healthy donors before adipose tissue donation.
Human umbilical cord blood samples were collected from healthy full-term births at the Shanghai Ninth People’s Hospital following IRB-approved protocols (IRB no. SH9H-2023-TK189-1), or purchased from the Shangdong Qilu Stemcell Engineering co., ltd. All blood samples were received as de-identified, therefore, the information on the age and/or gender of the donors is not available.
Cell and tissue culture
All cell and tissue culture operations were conducted under sterile conditions in laminar flow hoods. Cells or microfat tissues were maintained in an incubator at 37 °C and a constant 5% CO2 atmosphere.
For single cell expansion, SVFs or ADSCs were seeded at a density of 1 × 105 cells/cm2 in 100-mm dishes and cultured in α-MEM supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 1% PSG. The medium was changed twice per week. Prior to reaching confluence, cells were dissociated using 0.25% Trypsin-EDTA for passaging purposes. For microfat tissue culture and subsequent differentiation, RMF tissues or RMF pellets were cultured on ultralow attachment cell culture plates in specific medium, with the medium being changed twice per week.
Generation of RMF pellets from human adult adipose tissue
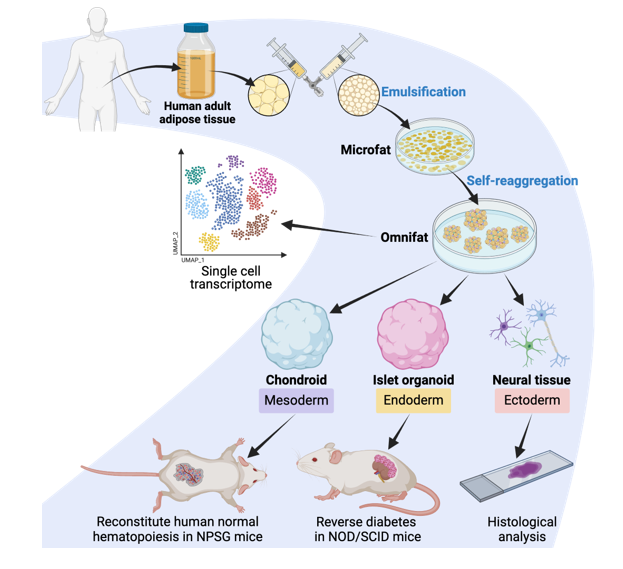
To generate chondroid pellets, 4-mm RMF pellets were generated through a conventional protocol as illustrated in Extended Data Fig. 1a. Adipose tissues, collected in the form of lipoaspirates obtained from healthy donors during liposuction surgeries, were washed, minced, and centrifuged at 1200 g for 3 minutes. The adipose tissues on the top layer were loaded into a 20-ml syringe connected to another syringe via a three-way connector with a filter hole size of 2.4 mm. Then, the adipose tissues were emulsified into microfat tissues by transferring the content between syringes for 30 cycles. The emulsified microfat tissues were subjected to further centrifugation at 1200 g for 3 minutes. After centrifugation, the microfat tissues were purified with a liquid part in the bottom and an oil part on the top. The purified microfat tissues were subsequently seeded onto ultralow attachment 6-well plates with each well containing 1.5 ml of microfat in growth medium, consisting of α-MEM supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 1% PSG, 10-5 M ascorbic acid, 10-7 M dexamethasone, 5 ng/ml FGF-2, and 10 ng/ml PDGF-BB. Following suspension culture for 3 weeks, the fractionated microfat tissues gradually self-reaggregated into RMF tissue constructs. To obtain uniform-sized RMF pellets measuring approximately 4 mm in diameter, biopsy punches with a diameter of 4 mm were employed to extract them from the RMF tissue constructs, which were then placed into 24-well ultralow attachment plates for an additional week of suspension culture.
To generate islet organoids, 1-mm RMF pellets with high cell density and small diameter were generated from a modified protocol as illustrated in Extended Data Fig. 5a. To increase cellular density, the washed and centrifuged adipose tissues were sequentially passed through a one-way connector with a filter hole size of 2.0 mm and another one-way connector with a filter hole size of 1.0 mm for 30 cycles, respectively. Subsequently, the emulsified microfat tissues underwent centrifugation at 1600 g for 3 minutes. Increasing the number of mechanical emulsification steps and applying higher centrifuge force facilitated the disruption and removal of more mature adipocytes, thereby augmenting cell density. The purified microfat tissues could self-reaggregate into RMF tissues comparable to those generated using the conventional protocol after a 2-week suspension culture period. Subsequently, 1-mm RMF pellets were obtained by punching the resulting RMF tissues with a 1-mm biopsy punch, which were subjected to an additional 2 weeks of suspension culture for further enhancement of cell density.
SVF cells extraction and culture
The SVFs were isolated from subcutaneous adipose tissues. The adipose tissues were washed, minced, and digested in 0.075% collagenase NB4 for 45 minutes to 1 hour at 37 °C on an orbital shaker. Subsequently, the suspension was centrifuged at 300 g for 10 minutes, and the resulting SVF pellet was rinsed once with PBS, resuspended in α-MEM supplemented with 10% FBS, and filtered through a 100-μm strainer. Next, red blood cell lysis buffer was added to remove erythrocytes followed by straining the cells again through a 70-μm strainer. Nucleated cells were counted and stored at -150 °C for further use. For single-cell expansion, SVFs were seeded at a density of 1 × 105 cells/cm2 in 100-mm dishes and cultured in α-MEM supplemented with 10% FBS and 1% PSG in an incubator at 37 °C and 5% CO2 with medium changed twice per week.
Osteogenic, adipogenic, and chondrogenic differentiation of SVFs and ADSCs
In vitro adipogenic and chondrogenic differentiation of cells and ADSCs were induced.
Adipogenic differentiation: cells were seeded in 6-well plates at a density of 5 × 103 cells/cm2 and expanded until reaching confluence before initiating differentiation.
For adipogenic differentiation, cells were cultured in adipogenic induction medium for 3 days followed by adipogenic maintenance medium for 1 day. This 4-day cycle was repeated 4 times, after which the cells were further cultured in the adipogenic maintenance medium for an additional week. The composition of adipogenic induction medium included high-glucose DMEM, 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 1% PSG, 10 μg/ml insulin, 10-6 M dexamethasone, 500 μM IBMX, and 100 μM indomethacin. The composition of the adipogenic maintenance medium consisted of high-glucose DMEM, 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 1% PSG, and 10 μg/ml insulin. Following adipogenic induction, the cells were washed and stained with Oil Red O staining kit for 15 minutes to visualize lipid droplet formation.
Osteogenic differentiation: cells were cultured in osteogenic induction medium for 3 weeks with medium changed twice per week. The osteogenic induction medium comprised high-glucose DMEM supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 1% PSG, 10-5 M ascorbic acid, 10-8 M dexamethasone, and 10-2 M β-glycerophosphate. After osteogenic induction, the cells were washed and stained with Alizarin Red solution for 10 minutes.
Chondrogenic differentiation: 5 × 105 cells were seeded onto a type I collagen-based cylindrical porous scaffold (4 mm in diameter, 1 mm in thick; Yierkang Bioengineering Co., Ltd., Beijing, China). The constructs were then cultured in chondrogenic medium for 4 weeks with medium changed twice per week. The chondrogenic medium is composed of high-glucose DMEM, 1.25 mg/ml HSA, 10 mM HEPES, 1 mM sodium pyruvate, 1% PSG, 10-5 M ascorbic acid, 10-7 M dexamethasone, 1% ITS, 0.47 mg/ml linoleic acid, 10 ng/ml TGF-β3, and 10 ng/ml BMP-6 for 4 weeks.
Differentiation of RMF pellets toward chondroid tissues using the two-stage endochondral priming protocol
Differentiations of RMF pellets toward chondroid pellets were performed using a four-stage induction protocol (Fig. 2a) using the following an endochondral ossification approach: stage 1 cartilaginous pellets and stage 2 hypertrophic cartilaginous pellets.
Medium preparation for endochondral priming
Stage 1 medium: High glucose DMEM + 1.25 mg/ml HAS + 10 mM HEPES + 1 mM sodium pyruvate + 1% PSG + 10-5 M ascorbic acid + 10-7 M dexamethasone + 1% ITS + 0.47 mg/ml linoleic acid + 10 ng/ml TGF-β3 + 10 ng/ml BMP-6
Stage 2 medium: High glucose DMEM + 1.25 mg/ml HAS + 10 mM HEPES + 1 mM sodium pyruvate + 1% PSG + 10-5 M ascorbic acid + 10-8 M dexamethasone + 10-2 M β-glycerophosphate.
Differentiation of RMF pellets towards chondroid pellets
The differentiation process was initiated 4 weeks later after self-reaggregation culture by changing to differentiation media. 4-mm RMF pellets were in ultralow attachment 24-well plates (1 pellet per well) in a humidified incubator at 37 °C and 5% CO2. Media changes were as follows.
Stage 1 (chondrogenic priming): RMF pellets were cultured in stage I chondrogenic priming medium for 4 weeks.
Stage 2 (hypertrophic induction): RMF pellets were changed to stage II hypertrophic induction medium and cultured for 2 weeks. The induction medium was changed twice per week.
Differentiation of RMF pellets toward islet organoids using the four-stage induction protocol
Differentiations of RMF pellets toward islet organoids were performed using a four-stage induction protocol (Fig. 4a) using the following formulations: stage 1 definitive endoderm, stage 2 panarctic progenitor, stage 3 endocrine progenitor, and stage 4 islet organoid.
Medium preparation for islet organoid induction
Basal medium 1: MCDB 131 + 8 mM D-(+)-Glucose + 14 mM NaHCO3 + 0.1% BSA + 2 mM GlutaMAX + 1% PSG.
Basal medium 2: MCDB 131 + 4 mM D-(+)-Glucose + 14 mM NaHCO3 + 0.1% BSA + 2 mM GlutaMAX + 1% PSG + 0.25 mM Vitamin C.
Basal medium 3: MCDB 131 + 2 mM D-(+)-Glucose + 24 mM NaHCO3 + 2% BSA + 2 mM GlutaMAX + 1% PSG + 0.25 mM Vitamin C + 1:50 ITS-X.
Basal medium 4: MCDB 131 + 20 mM D-(+)-Glucose + 24 mM NaHCO3 + 2% BSA + 2 mM GlutaMAX + 1% PSG + 0.25 mM Vitamin C + 1:50 ITS-X + 10 µg/ml Heparin.
Basal medium 5: MCDB 131 + 2 mM D-(+)-Glucose + 2% BSA + 2 mM GlutaMAX + 1% PSG + 10 µg/ml Heparin + 1% MEM nonessential amino acids
Differentiation of RMF pellets towards islet organoids
The differentiation process was initiated 4 weeks later after self-reaggregation culture by changing to differentiation media. 1-mm RMF pellets were cultured in ultralow attachment 48-well plates (1 pellet per well) in a humidified incubator at 37 °C and 5% CO2. Media changes were as follows.
Stage 1 (definitive endoderm): RMF pellets were exposed to basal medium 1 supplemented with 100 ng/ml Activin A, 3 μM CHIR99021 for 1 day, then changed to basal medium 1 supplemented with 100 ng/ml Activin A for 3 days.
Stage 2 (pancreatic progenitor): On day 5, RMF pellets were cultured in basal medium 2 supplemented with 50 ng/ml KGF for 2 days. For day 7 and day 8, RMF pellets were cultured in basal medium 3 supplemented with 50 ng/ml KGF, 200 nM LDN193189, 2 μM Retinoic Acid, 0.25 μM SANT1, 0.25 uM TPPB, and 0.3 mM Taurine. For day 9 to day 12, RMF pellets were cultured in basal medium 3 supplemented with 50 ng/ml KGF, 0.1 μM Retinoic Acid, 0.25 μM SANT1, 0.25 uM TPPB, 200 nM LDN193189, and 0.3 mM Taurine.
Stage 3 (endocrine progenitor): RMF pellets were then exposed to basal medium 4 supplemented with 10 μM ALK5i II, 20 ng/ml Betacellulin, 0.1 μM Retinoic Acid, 0.25 μM SANT1, 1 μM T3, 1 μM XXI, and 10 mM Nicotinamide for 7 days.
Stage 4 (islet organoid): On day 21, RMF pellets were cultured in basal medium 5 supplemented with 10 µg/ml ZnSO4·7H2O, 1:1000 Trace Elements A, and 1:1000 Trace Elements B for 7 to 14 days.
Differentiation of RMF pellets toward neural tissues using the two-stage induction protocol
Medium preparation for neural tissue differentiation
Differentiations of RMF pellets toward neural tissues were performed using a two-stage induction protocol (Extended Data Fig. 10a) using the following approach: stage 1 neural stem cells and stage 2 neuronal cells or neuroglial cells.
Stage 1 neural stem cell differentiation medium: Neurobasal™ Plus medium + 1% B27, 1% N2 + 20 ng/ml EGF + 20 ng/ml FGF-2.
Stage 2 neural differentiation medium: Neurobasal™ Plus medium + 1% N2 + 1% PSG + 5% horse serum + 0.5 μM ATRA + 20 ng/ml EGF + 20 ng/ml FGF-2. For neuronal differentiation, 10 ng/ml BDNF was added; for glial cell differentiation, 10 ng/ml PDGF-BB was added.
Differentiation of RMF pellets towards neural tissues
The differentiation process was initiated 4-weeks later after self-reaggregation culture by changing to differentiation media. 1-mm RMF pellets were cultured in were cultured on ultralow attachment 48-well plates (1 pellet per well) in a humidified incubator at 5% CO2 at 37 °C. Media changes were as follows.
Stage 1 neural stem cell differentiation: RMF pellets were cultured in stage 1 neural stem cell differentiation medium for 2 weeks.
Stage 2 neuronal or neuroglial differentiation: RMF pellets were exposed to stage 2 neuronal differentiation medium or glial differentiation medium for 2 weeks, respectively. The induction medium was changed twice per week.
Subcutaneous transplantation of RMF-chondroid pellets into NPSG mice
Immunodeficient NOD/LtJ-PrkdcscidIl2rgem1/Shjh (NPSG) female mice, aged 4 weeks, were obtained from Shanghai Jihui Laboratory. For subcutaneous transplantation, RMF-chondroid pellets (4 pellets per mouse) were transplanted under the back skin of mice. RMF-chondroid grafts were explanted at 2, 4, 6, 8, and 12 weeks post-transplantation.
Xenotransplantation of hUCB-derived CD34+ cells in NPSG mice
Primary xenotransplantation
The hUCB-CD34+ cells were isolated using a CD34 microbead kit, following the manufacturer’s instructions. At 8 weeks post implantation, 5 × 105 hUCB-CD34+ cells were pooled from a minimum of 6 donors (in total 15 donors, both females and males) and injected intravenously into NPSG mice 24 hours after sublethal irradiation (Redsource RS2000 XPro; 200 cGy). Mice were euthanized at 8 and 16 weeks post hUCB CD34+ cell transplantation. The femurs and hOssicles were collected for subsequent analysis of chimerism, self-renewal, and differentiation (Fig. 3a). The transplantation experiments were repeated 5 times with a cohort of 6 recipient mice (each mouse bearing 4 ossicles) per transplant.
Secondary xenotransplantation
After eight weeks of primary xenotransplantation of hUCB-CD34+ cells, the recipient mice were euthanized and the femurs and hOssicles were collected to isolate hCD34+ cells using MACS. The isolated hCD34+ cells were then plated onto cytokine-containing methylcellulose medium. For secondary xenotransplantation, a secondary batch of NPSG mice, aged 8-10 weeks without pre-established hOssicles, received sublethal irradiation (200 cGy) 24 hours prior to the secondary transplantation. Subsequently, 1 × 106 hCD34+ cells enriched from mouse femurs or hOssicles were injected intravenously into irradiated NPSG mice, respectively. Eight weeks after the secondary transplantation, the femurs were collected for subsequent chimerism, self-renewal, and differentiation analysis. The secondary transplantation experiments were repeated 3 times with a cohort of 6 recipient mice per transplant.
Kidney subcapsular transplantation of RMF-islet organoid in NOD/SCID mice.
Immunodeficient NOD.Cg-Prkdcscid/Shjh (NOD/SCID) mice, aged 8-10 weeks, were obtained from Shanghai Jihui Laboratory. For nondiabetic cohorts, RMF-islet organoids (40 to 48 pellets per mouse) were loaded into catheter and transplanted under the kidney capsule of mice. RMF-organoid grafts were explanted by nephrectomy for analysis graft survival and vascularization at 1, 2, and 4 weeks post-transplantation (Extended Data Fig. 7a).
For diabetic cohorts, mice were rendered diabetic after a single intraperitoneal injection of streptozotocin (STZ; 150 mg/kg body weight). Four days after STZ treatment, nonfasted blood glucose was measured from tail vein samples using a OneTouch glucometer. Seven days after STZ treatment, hyperglycaemic mice (blood glucose higher than 200 mg/dL) were selected for further transplantation studies. RMF-islet organoids (40 to 48 pellets per mouse) or undifferentiated RMF pellets (42 to 48 pellets per mouse, derived from the same donors of the islet organoid group) were transplanted under the kidney capsule of mice. The mice underwent sham operation were left as controls. Nonfasted blood glucose and body weight were routinely monitored up to 12 weeks after transplantation. At select times, intraperitoneal glucose tolerance tests were performed following standard protocol. Briefly, after fasting the mice for 16 hours overnight, mice were intraperitoneally injected with glucose (2 g/kg body weight), blood glucose measured were measured at 0, 30, 60, 90, and 120 minutes after glucose challenges. Serum samples before and 30 minutes after glucose challenge were also collected. Serum human insulin levels were quantified using a Human Ultrasensitive Insulin ELISA kit. At 12 weeks after transplantation, nephrectomy was performed to harvest the grafted tissues for section and endocrine markers staining or digested into single cells for 10× scRNA-seq. At 13 weeks after transplantation, mice were euthanized and the recipients’ main organs including pancreas, heart, brain, liver, kidney, spleen, and lung were harvested for gross anatomy and histological analysis (Fig. 5a).
In vitro glucose-stimulated insulin secretion
Islet organoids were washed twice in Krebs buffer (KRB) and pre-incubated in KRB containing low glucose (2 mM) for 2 hours at 37 °C to remove residual insulin. Islet organoids were then challenged with three sequential treatments of alternating low-high-low KRB containing glucose (high; 20 mM), followed by depolarization with KRB containing 30 mM KCl and low glucose (2 mM). Each treatment lasted 30 minutes, after which 100 μl of supernatant was collected and human insulin quantified using the Human Ultrasensitive Insulin ELISA. The islet organoids were enzymatically dissociated into single cells using TrypLE™ Express Enzyme and viable cells were counted using a Countess II (Thermo). Human insulin measurements were normalized by viable cell counts.
Cell proliferation assay
Duo to the heterogeneity of cell populations in SVFs, RMF cells, P0 ADSCs, and expanded ADSCs. Following several pilot studies, different amounts of cells were seeded in 96-well plates to ensure different samples achieved the same adherent cell numbers at 24 hours post cell seeding: 7000 cells per well for RMF cells, 9000 cells for SVF cells, 3000 cells for P0 ADSCs, and 5000 cells for expanded ADSCs. Six wells were used for each group, and the CCK-8 standard curve was fitted according to the measurement results. Moreover, cells were cultured in α-MEM supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 1% PSG for 24, 48, 72, and 96 hours. Then, the cells were incubated with a CCK-8 reagent for 2 hours at 37 °C and 5% CO2. Thereafter, the medium was harvested to measure the optical density values at 450 nm using a microplate reader. The absolute number of cells was calculated by standard curve and the cell proliferation curve was plotted.
SVF and ADSC colony-forming unit assay
To determine colony-forming efficiency, 1 × 104 cells were seeded on 100-mm cell culture plates and cultured in α-MEM supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 5 ng/ml FGF-2. After 2 weeks, cultures were rinsed with PBS, fixed with 4% PFA stained with Crystal Violet for 10 minutes and rinsed with tap water. Colonies were imaged and counted.
Hematopoietic colony-forming unit assay
For the colony-forming unit assays, hCD34+ cells were sorted from hOssicle and mouse femur bone marrow using MACS. Then, 1 × 103 hCD34+ cells were seeded into 1 ml MethoCult™ H4434 Classic medium and cultured at 37 °C and 5% CO2 for 12-14 days. Hematopoietic colonies (BFU-E, CFU-GM, and CFU-GEMM) were quantified according to the manufacturer’s instruction.
Flow cytometry
SVF and ADSC subpopulation analysis
SVFs and ADSCs were trypsinized, washed, and filtered through 40-μm cell strainers. Then cells were incubated with Zombie Aqua™ dye and blocked with Human TruStain FcX™ before staining with the following mix of fluorochrome-conjugated mouse anti-human monoclonal antibodies: CD31-Brilliant Violet 605TM (WM59), CD34-PE/Cyanine7 (581), CD45-PerCP/Cyanine5.5 (HI30), CD73-Brilliant Violet 421TM (AD2), CD90-APC/FireTM 750 (5E10), and CD146-APC (P1H12) for 30 minutes at 4 °C. All antibodies were used at a 1:100 dilution. Samples were acquired on a CytoFLEX LX (Beckman Coulter) then data was analyzed using FlowJo 10. Flow cytometry analysis data were plotted as a percentage of MSCs (gated as CD45-CD73+CD90+), endothelial progenitor cells (EPCs, gated as CD45-CD31+CD34+), and pericytes (gated as CD45-CD31-CD146+) distribution among Zombie Aqua™ dye-negative live cells.
Humanized ossicle and mouse femur bone marrow cell analysis
Mice were euthanized and the femurs and hOssicles were collected. Bone marrow cells from mouse femurs and explanted humanized ossicles were isolated by flushing mouse bones with 29G needles and by crushing ossicles with micro pestles, respectively, into tubes containing FACS buffer (DPBS + 2% FBS). Bone marrow cells were incubated with Red Blood Cell Lysis Buffer, stained with Zombie Aqua™ dye and blocked with Human TruStain FcX™. For mouse HSC analysis, bone marrow cells were then stained with the mix of anti-mouse Lineage cocktail-PE (145-2C11; RB6-8C5; RA3-6B2; Ter-119; M1/70), Sca-1-PE/Cyanine7 (D7), c-Kit-APC (2B8), CD135-Brilliant Violet 421TM (A2F10) and CD34-FITC (RAM34) for 30 minutes at 4 °C. For mouse HPC analysis, bone marrow cells were stained with the mix of anti-mouse Lineage cocktail-PE (145-2C11; RB6-8C5; RA3-6B2; Ter-119; M1/70), Sca-1-PE/Cyanine7 (D7), c-Kit-APC (2B8), CD34-FITC (RAM34), CD135-Brilliant Violet 421TM (A2F10), IL-7Rα-APC/Cyanine7 (A7R34), and CD16/32-PerCP/Cyanine5.5 (93) for 30 minutes at 4 °C. All antibodies were used at a 1:100 dilution.
Samples were acquired on a CytoFLEX LX then data was analyzed using FlowJo 10. Flow cytometry analysis data were plotted as a percentage of LT-HSC (gated as L-S+K+CD34-CD135-), ST-HSC (gated as L-S+K+CD34+CD135-), MPP (gated as L-S+K+CD34+CD135+), CLP (gated as Lin-CD135+IL-7Rα+), MEP (gated as L-S-K+CD34-CD16/32-), CMP (gated as L-S-K+CD34+CD16/32-), and GMP (gated as L-S-K+CD34+CD16/32+) distribution among Zombie Aqua™ dye-negative live cells.
Transplanted hUCB-CD34+ cell analysis
8 weeks and 16 weeks after xenotransplantation of hUCB-CD34+ cells, bone marrow cells were isolated from mouse femurs and explanted humanized ossicles, respectively, into tubes containing FACS buffer. After red blood cells lysis with Red Blood Cell Lysis Buffer, bone marrow cells were incubated with Zombie Aqua™ dye and blocked with Human TruStain FcX™. Monoclonal anti-mouse CD45-FITC (30-F11), anti-human CD45-Brilliant Violet 421TM (HI30) were used to distinguish human from mouse cells. Monoclonal mouse anti human CD34-APC (581), CD38-PE (HB-7), and a human lineage cocktail of antibodies including PE/Cyanine 7-conjugated mouse anti human CD2 (RPA-2.10), CD3 (HIT3a), CD4 (RPA-T4), CD7 (CD7-6B7), CD8a (RPA-T8), CD14 (HCD14), CD16 (3G8), CD19 (HIB19), CD20 (2H7), CD56 (5.1H11), and CD235ab (HIR2) were used to sort human HSPCs. Monoclonal mouse anti human CD3-PE/Cyanine 7 (SK7), CD19-APC (HIB19), and CD33-PE (WM53) were used to analyze human mature blood cells. All antibodies were used at a 1:100 dilution and incubated at 4° C for 30 minutes. Cells were acquired on a CytoFLEX LX then data was analyzed using FlowJo 10. Flow cytometry analysis data were plotted as a percentage of hHSCs (gated as human lineage-CD34+CD38-), hHPCs (gated as human lineage-CD34+CD38+), human T cells (gated as CD3+), human B cells (gated as CD3-CD19+CD33-), and human myeloid cells (gated as CD3-CD19-CD33+) distribution among donor-derived hCD45+ cells.
8 and 16 weeks after the secondary xenotransplantation of mouse femur or humanized ossicle-derived CD34+ cells, bone marrow cells from mouse femurs were isolated by flushing mouse bones with 29G needles. The method for processing and staining BM cells as well as strategies for analyzing human HSPCs and mature blood cells are the same as above.
Islet organoid cell analysis
Islet organoids were dissociated by TrypLE™ Express Enzyme at 37 °C for 15-20 minutes and filtered through 40-μm cell strainers for ensuring single-cell suspensions. Cells were incubated with eBioscience™ Fixable Viability Dye eFluor™ 780 and blocked with Human TruStain FcX™. Then cells were fixed and permeabilized with Foxp3/Transcription Factor Fixation/Permeabilization solution according to the manufacturer’s instruction. For Stage 2 analysis, cells were then stained with anti-human PDX-1-PE (658A5) and NKX6.1-Alexa Fluor 488 (R11-560) for 30 minutes at 4 °C. For Stage 3 and Stage 4 analysis, cells were stained with the mix of anti-human C-Peptide-Alexa Fluor 647 (U8-424) and PDX-1-Alexa Fluor 488 (658A5) or the mix of C-Peptide-Alexa Fluor 647 (U8-424) and NKX6.1-Alexa Fluor 488 (R11-560) for 30 minutes at 4 °C. All antibodies were used at a 1:50 dilution. Samples were acquired on a LSR Fortessa X-20 (BD Biosciences) then data was analyzed using FlowJo 10.
Live and dead cell analysis
RMF pellets with diameters of 1, 2, 4 mm were dissociated by 0.15% Collagenase NB4 at 37 °C for 30-40 minutes and filtered through 40-μm cell strainers for ensuring single-cell suspensions. For live and dead cell analysis, cells were incubated with eBioscience™ Fixable Viability Dye eFluor™780 for 30 minutes 4 °C. Samples were acquired on a CytoFLEX LX and then data was analyzed using FlowJo 10.
Magnetic-activated cell sorting MACS
Enrichment of CD34+ cells from hUCB and bone marrow
Mouse femurs and explanted ossicles were collected, crushed, flushed, and resuspended into MACS Buffer (DPBS, 0.5% BSA, 2 mM EDTA) and incubated with anti-human CD34 microbeads at 4 °C. After 30 minutes, the samples were rinsed and resuspended into MACS Buffer. The purification was done with the Mini & MidiMACS Starting Kit according to the manufacturer’s instruction.
Dead cell removal
Islet organoids were dissociated by TrypLE™ Express Enzyme at 37 °C for 15-20 minutes and filtered through 40-μm cell strainers for ensuring single-cell suspensions. Cells were washed and resuspended into binding buffer and incubated with Dead Cell Removal Microbeads for 15 minutes at room temperature. The purification was done with the Mini & MidiMACS Starting Kit according to the manufacturer’s instruction.
Mouse cell removal
Islet organoid grafts were explanted from the kidney and then dissociated by TrypLE™ Express Enzyme at 37 °C for 15-20 minutes and filtered through 40-μm cell strainers for ensuring single-cell suspensions. Cells were washed and resuspended into MACS Buffer and incubated with Mouse Cell Depletion Cocktail for 15 minutes at 4 °C. The purification was done with the Mini & MidiMACS Starting Kit according to the manufacturer’s instruction.
Tissue processing and histological analysis
Tissue samples were fixed in 4% PFA for 24 hours at room temperature. In vivo ossicle samples were further decalcified for 7 days in 0.5 mM EDTA, changing the solution twice per week. Next, samples were embedded in paraffin and cut with a microtome in 5-μm sections. The sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (HE), 0.1% Safranin O/Fast green solutions to detect glycosaminoglycans, Movat’s pentachrome staining kit, or leukocyte acid phosphatase kit for detecting tartrate-resistant acid phosphatase (TRAP) activity.
Immunofluorescence staining
For immunofluorescence staining, slices were blocked with blocking buffer for 1 hour at room temperature and incubated with primary antibodies with appropriate dilutions overnight at 4 °C, followed by washing in PBST (PBS + 0.1% Triton X-100), and incubation with secondary antibodies for 1 hour at room temperature. Nuclei were stained with DAPI for 10 minutes. Antibody details are provided in the key resources table. Representative images were taken using a Leica Stellaris 8 confocal microscope.
Immunohistochemical staining
For immunohistochemical staining, temperature-based (70 °C/160 W, 40 min) or enzymatic digestion-based antigen retrieval was performed at either high or low pH depending on the antibody. Then, following blocked with blocking buffer for 1 hour at room temperature and incubation with primary antibody or appropriate amount of IgG control overnight at 4 °C, slices were washed and incubated with biotinylated secondary antibodies. Chromagen development was achieved by incubating sections with DAB or Vector red according to manufacturer’s instructions. The slices were mounted and photographed using a Zeiss Axio Imager M2 microscope. Antibody details are provided in the key resources table.
Live and dead cell staining
The 1-mm, 2-mm, and 4-mm RMF pellets were stained with the LIVE/DEAD™ Viability/Cytotoxicity Assay Kit according to the manufacturer’s instruction and then washed in PBST. Then, the samples were scanned using a Leica Stellaris 8 confocal microscope.
Tartrate-resistant acid phosphatase (TRAP) staining
TRAP staining was performed in paraffin-embedded tissue sections using a leukocyte acid phosphatase kit. Briefly, deparaffinized slices were immersed in 0.2 M acetate buffer at pH 5.0 containing sodium acetate trihydrate and sodium tartrate for 20 minutes. The slices were then incubated in fresh, pre-warmed staining solution containing 1mg/ml Naphthol AS-MX phosphate powder and 1 mg/ml Fast Red TR Salt 1,5-naphthalenedisulfonate salt at 37 °C for approximately 2 hours. Upon visual confirmation of bright red color development, the slices were rinsed and subsequently counterstained with Mayer’s Hematoxylin solution for 1 minute. Slices were mounted and photographed using a Zeiss Axio Imager M2 microscope.
Confocal imaging, analysis, and quantification
Stained tissue and organoid slices were imaged on a Stellaris 8 confocal microscope or a Zeiss Axio Imager M2 microscope. TIFF files of the merged images were processed and analyzed for measurement of mean fluorescence intensity and manual counting of positive cells for any given markers using ImageJ software. For quantification of cell distribution, regions of interest (ROIs) were defined to divide the organoids in bins, and number of positive cells, mean fluorescence intensity, and area for any given ROI was determined. Similarly, for quantification of cell abundance, positive cells for each marker were manually counted and normalized over DAPI+ cells. Quantifications were performed across multiple regions and multiple organoids using at least n = 3 biological replicates. Quantification was performed by two independent investigators.
RNA isolation and reverse transcription (RT)-quantitative PCR (qPCR)
RNA isolation of primary cells, tissues, or organoids were performed following the manufacturer’s instructions. Briefly, total RNA was isolated using the SteadyPure Quick RNA Extraction Kit according to the manufacture’s instruction. Extracted RNA was reverse transcribed into cDNA using the Primerscript RT master kit. cDNA was stored at -20 °C or -70 °C for long time storage.
qPCR was prepared with TB Green Premix Ex Taq kit and detected by Applied Biosystems QuantStudio 3 (Thermo). Raw data were analyzed by ABI Prism 7000 SDS Software (Applied Biosystems). Comparative CT method (2-∆∆CT method) was chosen to evaluate differentially expressed genes. All relative expression levels were normalized to the housekeeping gene GAPDH. A list of the primer sequences for qPCR were listed in Table S2.
DNA content quantification
The DNA content of RMF tissues or pellets were quantified using an Animal Tissues/Cells Genomic DNA Extraction Kit according to the manufacturer’s instruction. The amount of DNA was detected by the NanoDrop Lite Plus Spectrophotometer (Thermo). The DNA content was calculated by dividing the amount of DNA of each RMF sample by the volume of samples.
Scanning electron microscopy (SEM)
Samples were fixed with 0.25% glutaraldehyde overnight at 4 °C and washed with PBS. The samples were gradually dehydrated with 30%, 50%, 70%, 90%, and 100% ethanol, coated with gold and imaged with a microscope (ZEISS GeminiSEM 300).
Transmission electron microscopy (TEM)
To analyze granular ultrastructure, islet organoids were fixed with a mixture containing 1.25% PFA, 2.5% glutaraldehyde, and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4) at 4 °C for overnight. Samples were then washed in 0.1 M cacodylate buffer and post fixed at room temperature with a mixture of 1% OsO4/1.5% KFeCN6 once for 2 hours then once for 1 hour. After washing with water, samples were stained in 1% aqueous uranyl acetate for 1 hour, washed, and subsequently dehydrated in gradual ethanol (30% to 100%) and propylene oxide. Then, samples were embedded in Epon 812. The samples were sectioned by diamond knife (Leica EM UC7) to 70-90nm, followed by stained with uranyl acetate and lead citrate for contrast. All the specimens were viewed by transmission electron microscope (HITACHI H-7650).
Microcomputed tomography (microCT)
Following fixation in PFA and storage in PBS, microCT data were acquired from explanted ossicles by using a high-resolution scanner (SkyScan1172, Skyscan, Belgium) and 0.5-mm aluminum filtered X-rays (applied voltage 50 kV; current, 200 mA). Transmission images were acquired during a 360° scan rotation with an incremental rotation step size of 0.25°. Reconstruction was performed using a modified Feldkamp algorithm at an isotropic voxel size of 2.5 μm. 3D rendering of the structures was performed using NRecon software (Bruker microCT, Kontich, Belgium) and CTvox (Bruker microCT, Kontich, Belgium) and analyzed by the program CTAn (Bruker microCT, Kontich, Belgium).
Single-cell sequencing and analysis
Single-cell sample preparation
The tissue samples were washed and digested using collagenase and incubated in a shaking water bath set at 37 °C for 60 minutes to facilitate optimal cell dissociation. Subsequently, debris and dead cell removal was performed to eliminate any remaining tissue fragments and acquire a single-cell suspension with high cell viability. Then, cell count and viability was estimated using fluorescence Cell Analyzer (Countess II, Thermo) with AO/PI reagent. Finally, the harvested cells were washed twice with PBS and then resuspended in PBS with 0.04% BSA at a density of 1 × 106 cells per ml.
Single-cell RNA-seq library construction and sequencing
Single-cell RNA-Seq libraries were prepared using a 10× Chromium Next Gel Bead-in-Emulsion (GEM) Single Cell 3’ Reagent Kits v3.1 from 10× Genomics as instructions. Briefly, cells were mixed with reverse transcription reagent and then loaded into the sample well in Chromium Next GEM Chip G. Subsequently Gel Beads and Partitioning Oil were dispensed into corresponding wells separately. After emulsion droplet were formed, reverse transcription was performed. The resulting cDNA was then purified, amplified, cleaned, and ligated to sequencing adaptors. Finally, the indexed PCR was employed to amplify the DNA fragments. The indexed sequencing libraries were cleared with SPRI beads, quantified by qPCR, and ultimately sequenced on a NovaSeq 6000 platform (Illumina) with PE150 read length.
Single-cell RNA sequencing data processing
The Cell Ranger Single-cell Software Suite61 was used to perform sample demultiplexing, barcode processing, and single-cell 3ʹ gene counting. First, UMI tags and barcode sequences were extracted from Read1, followed by alignment of Read2 (containing the cDNA insert) to a reference genome using STAR62. Next, Barcodes and UMIs were filtered based on quality. Last, PCR duplicates were identified by shared barcode sequences, UMI tags, and gene IDs. Confidently mapped non-PCR duplicates with valid barcodes and UMIs were utilized to generate the gene-barcode matrix. Cell barcodes were determined, distinguishing between cells and empty partitions. Meaningful read counts were calculated based on valid barcodes, UMIs, and mapped reads.
Single-cell RNA transcriptome analysis
The clustering and visualization were finished by Python with methods in the package Scanpy (v1.10.1)63. Quality control measures included removing potential doublets with the Scrublet software (v0.2.3)64 and filtering out cells with fewer than 2,500 UMIs or fewer than 1,000 genes per cell. Additionally, genes detected in fewer than three cells were excluded. The counts per cell were normalized to reach a total of 10,000 counts per cell and then subjected to a logarithmic transformation. Cells were grouped into clusters by identifying highly variable genes. Subsequently, adjustments were made for total UMI count per cell and expression of mitochondrial genes through regression analysis, followed by standardization of the resulting residuals. Principal component analysis was employed to reduce dimensionality to 50 principal components. A nearest neighbor graph was constructed based on these components, enabling the application of Leiden clustering to segregate cells into distinct clusters.
Ternary plot was used to position cells based on the proportions of signature genes expressed in native fat tissue, RMF pellets, and expanded ADSCs. The MDS plot was created using the vcd package with quantile normalized log2-CPM values. The log2-CPM values for each gene across cells were calculated with a prior count of 1, using the cpkm function from edgeR.
Trajectory analysis was carried out using Slingshot (v1.2.0)65. After selecting the subpopulations, the pseudotime values were calculated using slingshot. Then, genes associated with the pseudotime direction were calculated using the ‘associationTest’ in tradeSeq. Finally, the gene list was filtered for plotting based on p-value, meanLogFC, and whether the genes are differential between subpopulations.
The ‘AddModuleScore’ function from the Seurat (v4.4.0)66 package was employed to calculate gene expression module scores for each cell. Cells within the same cluster showed comparable module scores, indicating similar gene expression profiles and likely analogous cellular functions or states. The signal distribution within each cluster was analyzed and visualized using violin plots with the ‘VlnPlot’ function in Seurat.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as means ± SD and were analyzed by GraphPad Prism 9 (GraphPad Software). Paired or unpaired two-tailed Student’s t tests were performed when two groups of samples were compared. One-way or two-way ANOVA with Tukey’s or Dunnett’s tests were performed when multiple groups were compared. Log-rank (Mantel-Cox) test is used for comparison of survival rate between multiple groups. p values are indicated in the figures as: ns; p > 0.05; ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001; ∗∗∗∗ p < 0.0001. Statistical details for each experiment can be found in the figures and the legends.
{kind=link}