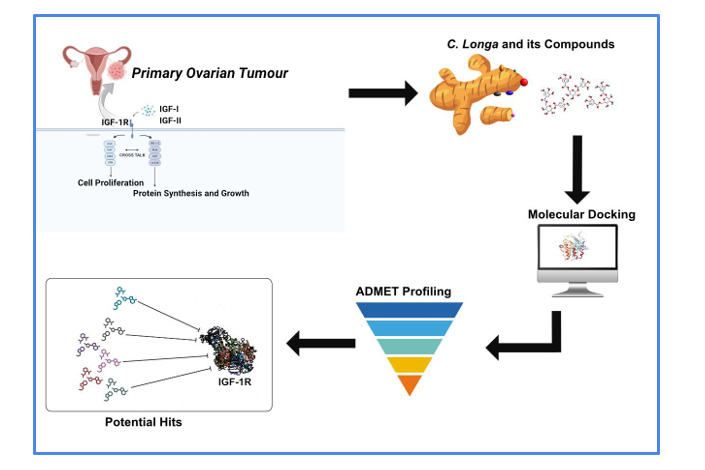
Molecular docking simulations provide us abilities to study protein-protein interactions as well as protein-ligand interactions with varieties of computational softwares available for these docking studies in drug discovery. These interactions are studied by predictions of binding sites which are determined by using the shape, electrostatic interactions, van der Waals interactions, hydrogen bond formation and coulombic interactions to study the ligand and protein receptors or enzyme interactions[17]. Over 60 available programs are used for docking studies, AutoDock Vina, GOLD, FlexX and MoE-Dock are just a few examples[18]. Our study used the Glide docking tool in Maestro 11.1 to predict binding affinities of C. Longa compounds with the IGF-1R protein receptor. We screened twenty-four phytochemical compounds virtually from the phenolic extracts of C. Longa rhizome and identified four lead compounds based on their docking scores. Ligands are ranked lead compounds based on their binding affinity scores, the higher the binding affinity scores, the more potential it has to bind to the target protein and this score is retrieved in the process of post docking analysis[17]. The lead compounds identified in this study were digalloyl-hexoside, hyperoside, valoneic acid dilactone, andcaffeic acid. Digalloyl-hexoside exhibited the highest negative docking score (-8.849 kcal/mol; Table 1) followed by close number of score for hyperoside and valoneic acid dilactone (-8.843 kcal/mol, -8.472 kcal/mol, respectively). Hyperoside and valoneic acid dilactone were also predicted to be candidates to inhibit the IGF pathway with docking scores. Caffeic acid showed the lowest docking score with -7.985 kcal/mol indicating a least binding affinity with IGF-1R. The three ligands’ molecules digalloyl-hexoside, hyperoside and valoneic acid dilactone showed ability to interact with the target receptor and form both hydrogen and other electrostatic/hydrophobic interactions with several amino residues. Digalloyl-hexoside, hyperoside, and valoneic acid bilactone showed six hydrogens bonds while caffeic acid had three hydrogen bonds (Table 1). Valoneic acid dilactone compound exhibited the highest active site with 7 residues followed by Hyperoside with 5 residues, Digalloyl-hexoside with 3 residues, and at the last Caffeic Acid with 1 residue (Table 1; Figure 1). The active site was ASP 1056, LYS 1003, and GLU 1050. Among the active residues involved, polar amino acids were highly dominant, which can significantly influence key structural parameters such as hydrophobicity, solvent accessibility, and secondary structure. This indicates that digalloyl-hexoside has a great affinity with a favorable interaction with IGF-1R. Previous studies have highlighted the anticancer properties of identified C. longa compounds, particularly in the context of inhibiting cancer cell proliferation. Prominently, digalloyl-hexoside, from gallic acid demonstrated considerable anticancer activity through multiple pathways [19]. Methanolic extract from the compounds of C. Longa rhizomes have been shown to inhibit the growth of the human hepatoma cell line (HepG2), this study identified lead compounds that include ar-turmerone (20.50 %), β-sesquiphellandrene (5.20 %) and curcumenol (5.11 %), and Curcumin[20]. Consequently, these lead compounds provide insight to building a strong folding and stable configuration during binding with target proteins and in the case of our study, IGF-1R [21,22,23].
Pharmacokinetics profile of drugs include studying the Absorption, Distribution, Metabolism and Excretion (ADME) properties of drug candidates. This is a very important stage in drug discovery and development as it helps to look into cost, efficacy, toxicity and treatment strategies for patients as drugs that do not meet up to a standard pharmacokinetic profile, have a higher tendency to result in failures in clinical trials[24]. Toxicity of the drug was estimated by the degree of drug-drug interactions[25]. Digalloyl-hexoside, Hyperoside, Valoneic acid bilactone compounds showed low gastrointestinal absorption while caffeic acid had the highest absorption rate. While the first three compounds exhibited low metabolic rates, negatively affecting their bioavailability and limiting their ability to reach the target site through the bloodstream, caffeic acid displayed significantly improved pharmacokinetic properties. Specifically, caffeic acid demonstrated a high absorption rate and efficient metabolism, indicating its superior capacity to reach and act upon the target site. Moreover, its remarkable skin permeability (with a value of -6.58) suggests that caffeic acid is particularly well-suited for topical or transdermal delivery, offering an effective method of permeating the skin. Ideally, none of the molecules showed any penetration through the blood-brain barrier. Digalloyl-hexoside, Hyperoside, Valoneic acid bilactone, and caffeic acid all showed low oral bioavailability within the range of 0.11-0.56, making them ineffective via oral use (table 2). Results from the interactions of our lead compounds with the five cytochrome P450 families of enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4) showed that extracted compounds have no inhibitory properties on these enzymes which indicates that there is no impact or alteration on the normal metabolic processing of other drugs.
The Lipinski rule of five is one of the rules providing standards for compounds to be identified as drug candidates.The Lipinski rule of five considers five parameters which includes the molecular weight, polar surface area, lipophilicity (Log P), hydrogen bonding, and charge[26]. Our study also evaluated the lead compounds based on the Lipinski rule of five to screen out compounds with probable absorption problems. Caffeic acid showed no violation of the Lipinski rule of five, indicating that it is suitable for oral administration. According to the rule, poor absorption or permeation of a drug is more probable when the chemical structure fulfills two or more of the following criteria: the Molecular weight (MW) is greater than 500, the calculated log P value is above five, there are more than five hydrogen bond donors (–NH–, –OH), the number of hydrogen bond acceptors (–N=, –O–) is greater than ten [23]. Lipinski’s rule was tested, caffeic acid complied with the Lipinski rule of five while the other compounds had two violations. It has been demonstrated that heavier molecules have a lower probability of absorption and site-specific localization [24]. All lead compounds showed molecular weights below 500g/mol, this is significant in that it increases the likelihood that they will be easily absorbed by the body since smaller molecules penetrates the cell membranes more easily and are less likely to be substrates for efflux pumps and metabolic enzymes that can reduce a drug's bioavailability. For Oral absorption and water solubility Log P and LogS were evaluated. Molecules showed acceptable results for the former test with Hyperoside as the highest with 2.11, while less water solubility was indicated by the latter test as all results were than -3.0. Furthermore, membrane permeability and the movement of molecules were measured as Caffeic acid showed the lowest number among them. The Hyperoside, Digalloyl-hexoside, and Valoneic acid bilactone molecules showed higher values, while caffeic acid had 77.76 polar surface area (PSA) value was the highest efficient membrane permeability, and this was predicted to be directly linked with the molar mass of the modules.
LIMITATIONS
Our study focused on only the bioactive compounds in the C. Longa rhizome which limits us to just twenty-four phytochemical compounds present in C. longa rhizome to be screened for identification of the ligands and this represents a small portion of the copious bioactive compounds present in the entire plant. Additionally, Molecular Mechanics Generalized Born Surface Area (MMGBSA) which serves to estimate the binding free energy of the ligands and predict the strength of non-covalent interactions between ligands and protein (receptor) was not determined for the ligands in our study, also, wet lab experiments were not carried out to validate the finding of our studies due to funding issues.
{kind=link}