The prevalence of CS in Japan is 2.5 per million(Wu et al., 2020). Although the incidence of the disease is low, the prognosis is poor and there is no medicine to treat or cure it. Symptomatic treatment and avoidance of sunlight exposure are the only ways to prolong life. Studies have shown that the variation spectrum of ERCC8 in Chinese is different from that in other populations. Exon 4 rearrangement is the main variation in CSA patients of Chinese Han nationality(Xiaozhu Wang et al., 2017). To date, the HGMD Professional database has revealed 96 variations in the ERCC8 gene, missense/nonsense variations (39.6%)、splicing variations (22.9%)、small deletions (16.7%)、small insertions (5.2%)、small indels (3.1%)、gross deletions (10.4%)and complex rearrangements (2.1%). The clinical symptoms of patients with nonsense and frameshift variations may be more typical, while the clinical symptoms of patients with missense variations are less pronounced. These results suggest the heterogeneity of CS phenotype(X. Wang et al., 2023).
ERCC8 is located on 5q12.1 and consists of 12 exons, encoding a CSA protein containing 396 amino acids. The protein contains seven highly conserved repeat WD40 domain motifs (short ~ 40 amino acids residues often terminating in a Trp-Asp (W-D) dipeptide)(Gauhar et al., 2022). The CSA protein mediates an important DNA repair pathway, nucleotide excision repair (NER), a DNA repair system present in a various species to remove DNA damage. NER consists of global genome NER (GGNER) and transcription-coupled NER (TCNER). TCNER is initiated after RNA Pol II recognizes transcriptional damage, which not only effectively prevent transcription at the damaged site but also recruits ERCC8 and ERCC6. Deficiency in these two genes can induce the occurrence of CS and UV-sensitive syndrome(Crochemore et al., 2023; Cui et al., 2015; Liu et al., 2024) .
In this study, the proband exhibited a chronic onset type with gradual progression, showing typical clinical manifestations of CS. Laboratory tests revealed multiple organ damage, brain atrophy, and decreased white matter volume, clinically consistent with Cockayne syndrome. At the age of 3, she underwent whole exome sequencing for a diagnosis of "developmental delay," but the results were negative. The unusual phenotype and limited phenotype keywords made it challenging to identify pathogenic variants. Additionally, filtering out synonymous variants and suspicious single exon deletion contributed to the negative outcome of the whole exome sequencing.
To confirm the diagnosis, further genetic testing was performed. Whole exome sequencing revealed that the proband carried the ERCC8 gene c.1041G > A (p. Gln347Gln) heterozygous variation and a suspected heterozygous deletion in exon 1. Sanger sequencing showed that the heterozygous variation of c.1041G>A was inherited from the mother, who had a normal phenotype, and was an unreported variation. Quantitative real-time PCR was used to rule out the possibility of false positives for the exon 1 deletion and confirmed that the variation came from a father with a normal phenotype, consistent with the autosomal recessive inheritance model. According to the ACMG guidelines, c.1041G > A (PM2_supporting + PM3 + PP3 + PP4) was rated as a variant of uncertain significance, while exon 1 deletion (PVS1 + PM2_supporting + PP4) was classified as a pathogenic variation.
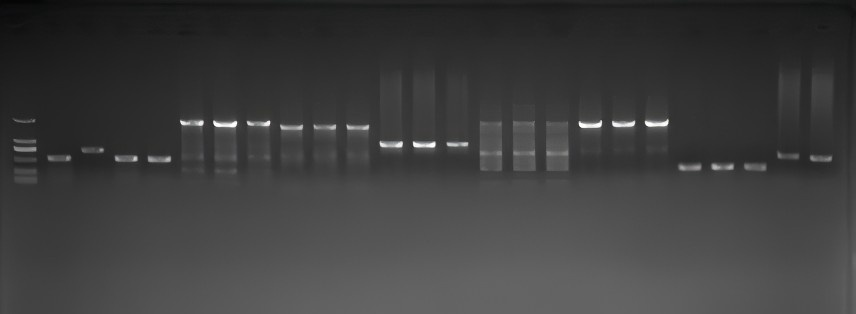
The c.1041G>A variation is located at the junction of exon 10 and intron 10, potentially affecting splicing and causing disease. Mutation taster (Disease_causing, predicted value of 1.0) and CADD software (predicted value of 33) predicted it as harmful. The SSP score was 0.93, indicating a high probability of splicing site activation. PCR electrophoresis showed that the amplified product of ERCC8 cDNA in the subjects was about 198 bp shorter than the 716 bp product of the normal control, matching the length of exon 10. RNA high-throughput sequencing suggested significant absent of exon 10 at the transcriptional level. Therefore, we believe that the c.1041G>A synonymous variation affects splicing, resulting in exon 10 deletion. According to the ACMG guidelines, c.1041G > A (p. 282_347del) (PVS1 + PM2_supporting + PM3 + PP4) was upgraded to a pathogenic variation, suggesting that even synonymous mutations with unknown significance can be highly pathogenic.
It is worth noting that transcriptome sequencing ERCC8 exon 1 did not reveal a deletion. Combined with the Quantitative real-time PCR, we believe that the exon 1 deletion may prevent the ERCC8 allele from initiating transcription, thus failing to transcribe and reverse transcribe into cDNA for sequencing. In addition, in actual RNA high-throughput sequencing, only one strand of mRNA exists, producing one strand of cDNA for testing after reverse transcription. Due to nucleic acid stability issues, a second cDNA strand needs to be synthesized, but we believe only one cDNA strand is detected. Therefore, the tested cDNA strand likely did not undergo deletion, showing no obvious deletion. Alternative splicing results indicated slightly lower mRNA expression of exon 1, but various factors affect mRNA expression. Consequently, it is unclear if this was due to a decreased copy number, resulting in lower exon 1 expression at the transcriptional level.
In short, the patient 's two ERCC8 alleles are affected as follows: one allele has exon 1 deletion preventing the initiation of the translation and transcription program, thus inhibiting normal protein expression; the other allele has a synonymous variation leding to the deletion of exon 10.
In this study, we used two different RNA sequencing methods and obtained consistent results: ERCC8 exon 10 deletion. Transcriptome sequencing analyzes the total RNA transcribed in a specific functional state, including mRNA and non-coding RNA. It allows for the study of gene function and structure at an overall level without the need for pre-designed probes for known sequences. This method provides more accurate digital signals, higher detection throughput, and a wider detection range, though the error rate is relatively high. Therefore, despite RNA Sanger sequencing being cumbersome, time-consuming, with lower throughput, longer read lengths, and higher costs, it remains the gold standard for diagnosis. Consequently, we combined both methods to achieve the most accurate results.
At the same time, it should be noted that the patient's disease is progressive, and the early stage was not diagnosed in time. The patient was admitted to the hospital at the age of 1 year and 7 months, with no obvious abnormalities in the head MRI results. At 3 years and 8 months, it was observed that white matter myelination was significantly delayed compared to children of the same age. By the age of 7 years, upon re-admission to the hospital, the patient had developed brain atrophy and low white matter mass. The final diagnosis was made based on comprehensive laboratory examination results and clinical phenotype parallel genetic diagnosis.
In 1970, Moosa and Dubowitz proposed that CS is a form of leukodystrophy(Moosa A, 1970). Most subseqnent case reports supported their view. In 2010(Koob et al., 2010), Koob conducted a retrospective analysis of 19 patients and summarized the main neuroimaging manifestations of CS disease as myelin dysplasia, calcification and brain atrophy. Early-onset patients exhibited more severe myelin dysplasia and significant calcification, while late-onset patients had less brain atrophy. These comprehensive neuroimaging findings are useful for the differential diagnosis of CS, distinguishing it from other childhood leukoencephalopathies and/or brain calcification. Baer et al. later screened 185 patient data from 135 articles published between 1988 and 2020(Baer et al., 2020), finding that 67% of patients showed ventricular enlargement, 76% had white matter abnormalities, 57% had posterior fossa abnormalities, and 41% had cerebral calcification. Using “Cockayne Syndrome” and “ERCC8” as keywords, we retrieved 25 articles from 2020 to 2024, encompassing 33 cases. Among these, 79.2% of patients showed brain atrophy, 33.3% showed brain calcification, 25% had white matter abnormalities, and 37.5% had myelin abnormalities. In our case, the patient also exhibited brain atrophy, hypomyelination, and decreased white matter volume, consistent with previous research findings.
{kind=link}