The necessity of an appropriate assembly strategy for mitogenomes
Genomic researchers have used different strategies for mitogenome analysis, including de novo assembly, mapping to reference genomes, and seed-mapping reads, and these have produced mitogenome sequences of variable quality. A survey of NCBI mitogenome data showed that, in some assemblies, MTPTs and repeat sequences were unfeasibly long compared with other individuals of the same species or close relatives of the same genus (Fig. 1). Although some differences among individuals of the same species have been observed, these differences are minimal for most species. The unusually long repeat sequences in some assemblies are also unlikely given the error-prone nature of MTPT and repeat assembly, which increases the chance of misassemblies. However, unfortunately, in most studies, mtDNA content was not conserved and misassemblies were undetectable, including for ribosomal protein genes and succinate dehydrogenase subunit genes sdh3 and sdh4. Some poorly assembled mitogenomes included artificial structures, such as inappropriate circularization, or had missing sequences, such as absence of ribosomal RNA genes rrnS and rrnL. In addition, assembly difficulties prompted some researchers to focus on analysis of fragmentary sequences without producing complete assemblies. The full scope of mitogenome evolution remains obscure in these cases, and it is challenging to reuse data when mitogenomes have been assembled and analyzed with a variety of methods. Improved assembly methods are needed to address the issues of existing assemblies.
Improvement of assembly
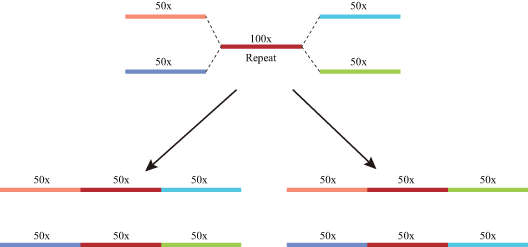
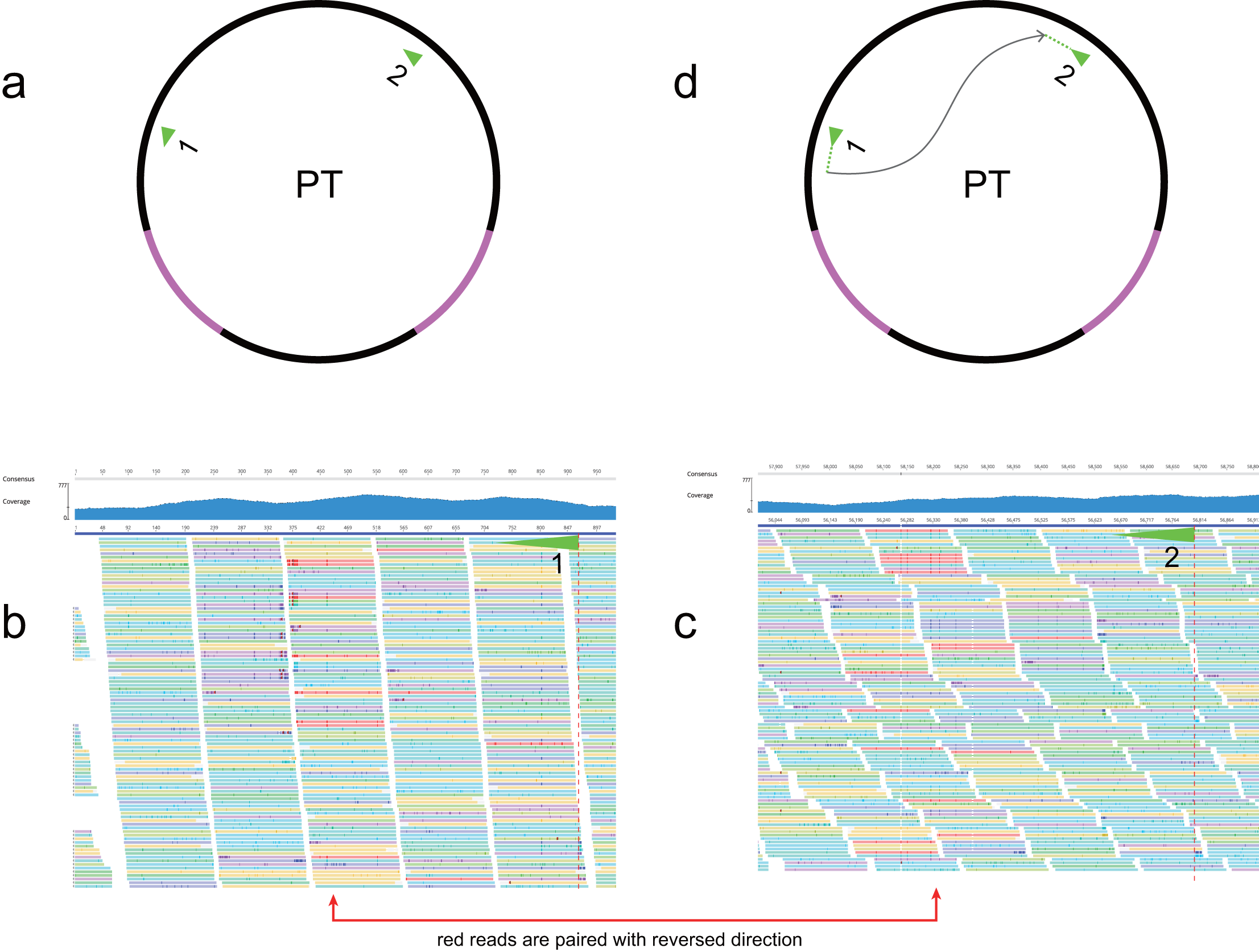
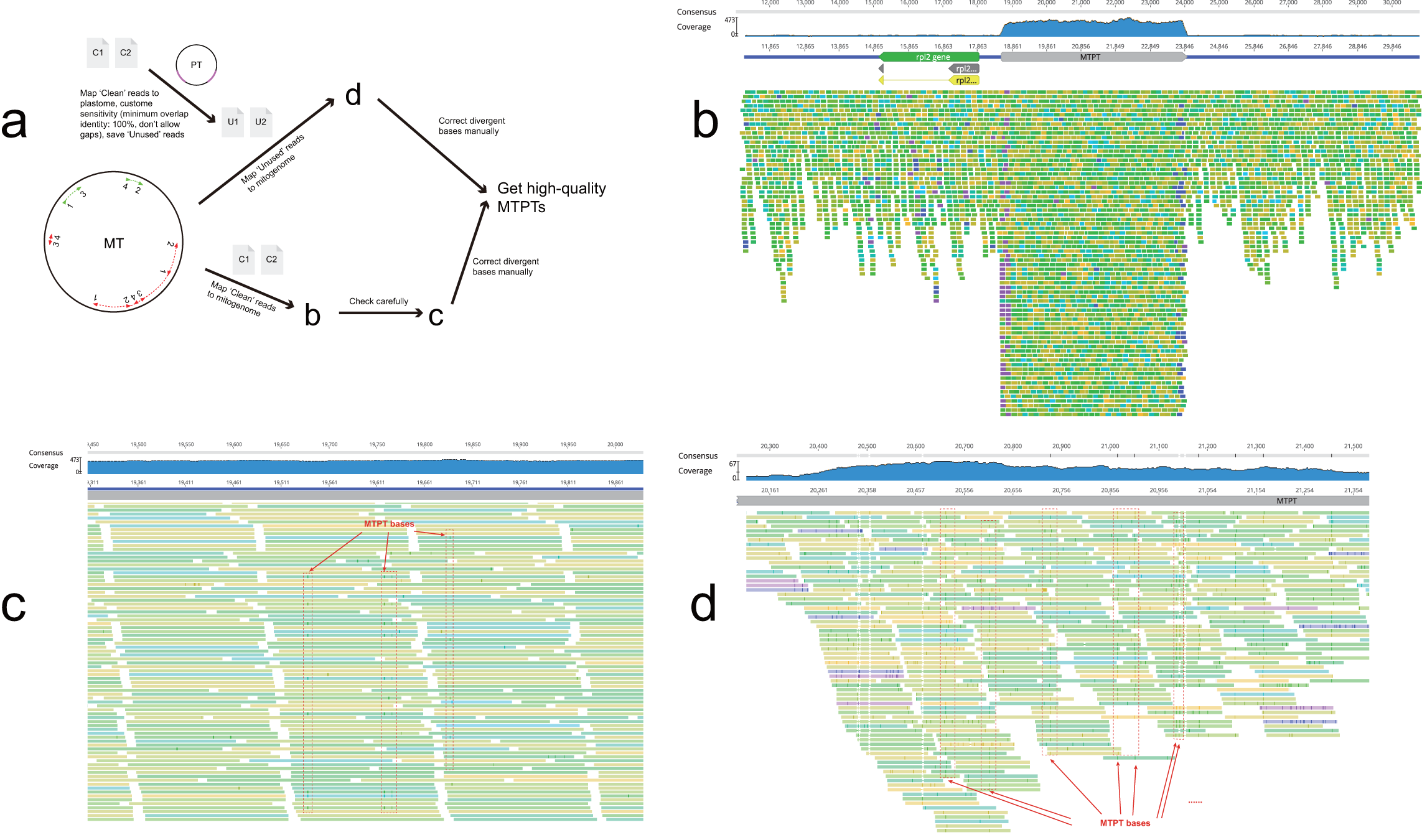
In most genome sequencing projects, total DNA is directly sequenced without separation of organellar and nuclear material. At present, mitochondrial sequence reads cannot be easily distinguished in whole-genome datasets. In our novel strategy, initial de novo assembly was followed by identification of mitochondrial contigs through gene identification and sequence coverage. Experience from our previous work and published mitogenomes suggests that angiosperm mitogenomes usually lack AT-rich regions so the read coverage is generally balanced. Mitochondrial contig ends occur when repeat sequences or MTPTs are encountered during assembly, i.e., contigs usually end either with repeats (repetitive ends) or MTPTs (MTPT ends). Our strategic workflow is outlined in Fig. 2. First, sequencing coverage was used to resolve repeat ends. Next, all MTPT ends were mapped to plastome and the highest numbers of potential connections were identified using their positions and directions. Circular mitogenomes were produced once all repetitive and MTPT ends were resolved. Finally, clean reads were mapped to the correct MTPTs (Fig. S3) and the final assembly.
Assembly results and completeness assessment
Our mitogenome assembly approach focused on solving issues caused by repeat and MTPT sequences in 23 Fagales species. Sequences of 2–3 Gb in size were used and coverage depth was 33–174. Of the 23 species, 13 yielded one or more circular mitogenomes, and the remaining 10 species contained one or more linear chromosomes (Table 1). Mitogenomes housed in multiple circular chromosomes did not share long repeats between the structures. In principle, circular chromosome assemblies can be achieved if all the repetitive and plastid sequence ends are connected. Sometimes, however, repetitive ends appeared unstable, with decreasing coverage towards the ends, or the paired end for an MTPT end could not be found. In these cases, ends could not then be connected properly (Fig. S4; Table S2). The sequencing datasets were derived from several different studies, and assemblies might therefore have been affected by the sequencing and library type, and read and insert lengths. For example, coverage of B. platyphylla and Lithocarpus, both produced by an Illumina Genome Analyzer, was much lower in some regions than others.
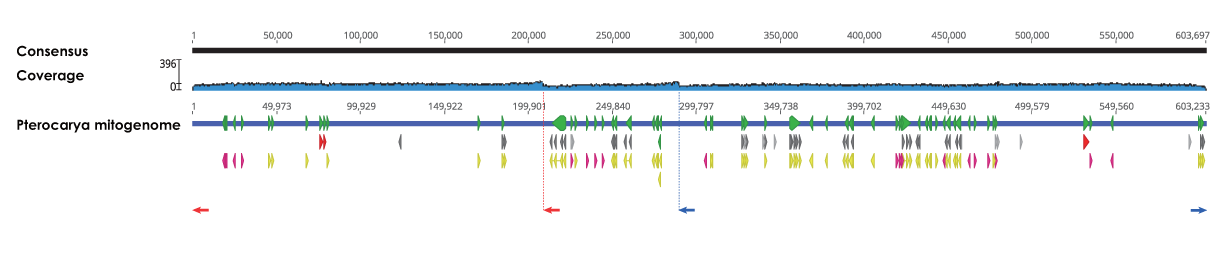
During this study, the mitogenome of Fagus sylvatica was published. Long and short reads were used to produce an assembly with a single circular chromosome of 504,715 bp in length (Mader et al., 2020). The sequence content of the published assembly was almost identical to that of the F. sylvatica assembly produced in this study, differing only in two bases (differences between individuals rather than differences in assembly). The only disparity between the two assemblies was an inversion of a sequence located between 900 bp repeats (Fig. S1). The repeat region was much longer than the insert length (450 bp; Table S1), and this inversion was therefore not unexpected. The consistency between our assembly and that of the previous study provided support for the practicability and reliability of our assembly methods. Furthermore, the mitogenomes in the two independent F. sylvatica projects were almost identical, indicating preservation of mitogenomes among individuals in at least some plant species.
Mitogenome size and content
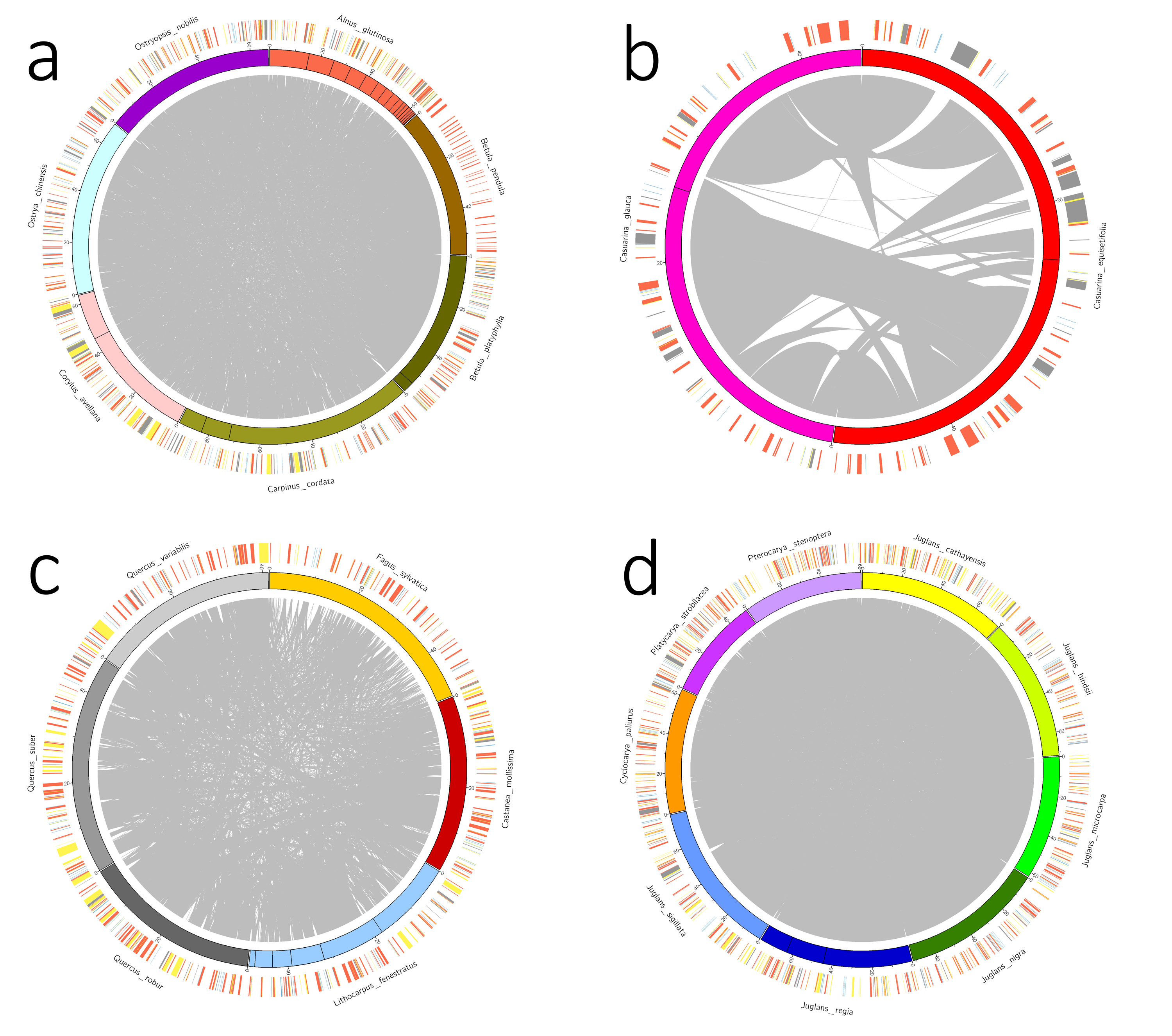
Characteristics of the mitogenome assemblies produced in this study, as well as previously published B. pendula and Q. variabilis assemblies, are provided in Table 1. Mitogenome sizes in Casuarinaceae, Fagaceae, and Myricaceae resembled those of distant relatives from Rosales or Fabales (400 Kb and 480 Kb on average, respectively, NCBI data). By contrast, mitogenome sizes were substantially expanded in Betulaceae and Juglandaceae. The largest mitogenome (922 Kb) was found in Carpinus (Betulaceae), and was much larger than those of confamiliar species. DNA content of mitogenomes did not differ substantially within families, but structures were often highly rearranged (Fig. S5). Mitogenome sequences were less similar between families, with some sequences having no homologs in other families (Fig. 3). The proportion of repeats in Fagales mitogenomes was small, normally less than 3% and no more than 6.2% of the total mitogenome length (Table 1). In Betulaceae, short repeats of less than 200 bp were more apparent, especially in Alnus (Table S3). MTPT percentages were also low, with only two species having more than 6% (Casu. equisetifolia, 13.5%; and Corylus, approximately 9.5%).
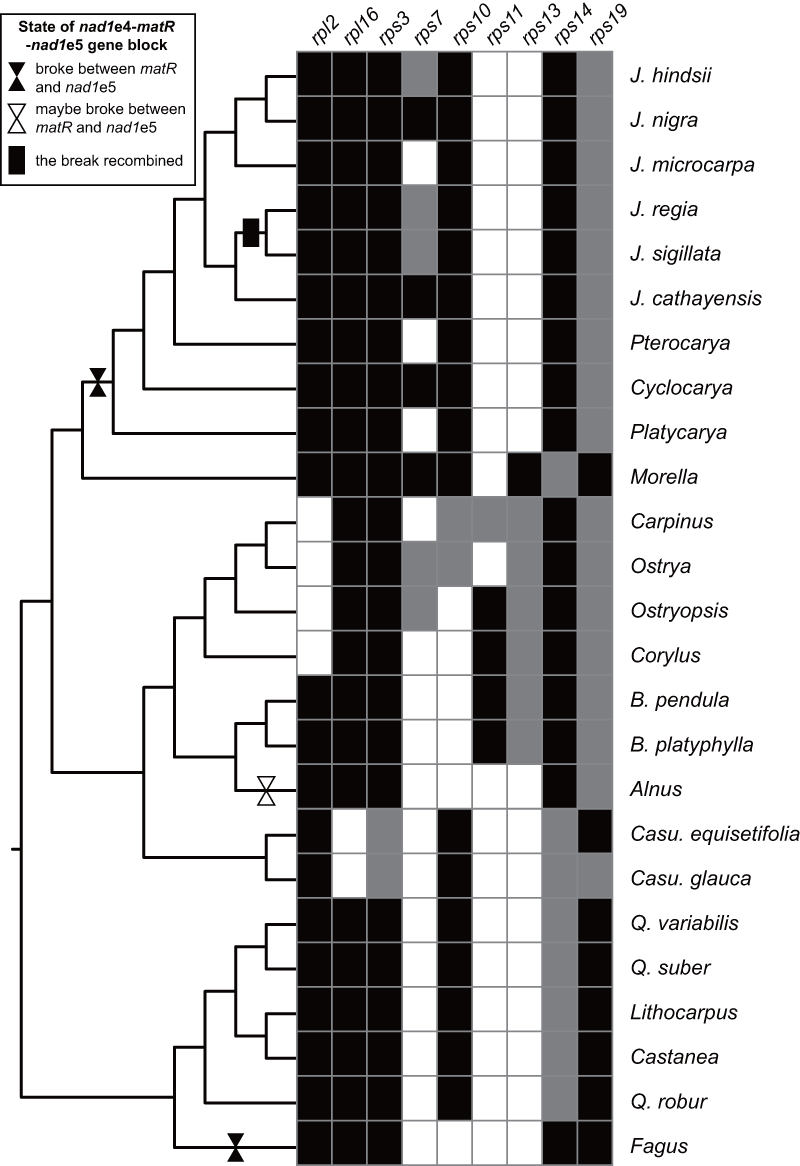
Conservation of some ribosomal protein genes was poor (Fig. S6), as in many plant species. Five of the seven Betulaceae species had rps11 sequences with approximate identities of 100%. Comparison of Betulaceae rps11 sequences with those in the NCBI nr database indicated similarities with rps11 in monocots or basal core angiosperms such as Triantha glutinosa (KX808303, Alismatales) and Liriodendron tulipifera (NC_021152, Magnoliales), consistent with previous research (Bergthorsson et al., 2003). These similarities suggested that HGT of rps11 may have occurred in a common Betulaceae ancestor, followed by differential losses in some species. Exon 4 of nad1 (nad1e4), matR, and nad1e5 form a colinear block in most angiosperms. This block was disrupted between matR and nad1e5 at least twice in Fagales species but, surprisingly, was recovered in Juglans sigillata and J. regia (Fig. S6).
Identification of a mitochondrial plasmid
A small circular mitochondrial plasmid, 2,888 bp in length, was found in Carpinus. Sequencing coverage was similar to that of the mitogenome. Plasmid GC content was 37.6%, which was much lower than normal mtDNA (Table 1) but was similar to Carpinus nuclear genomes (Car. fangiana: 37.6%, Yang et al., 2020). With the exception of a small 240 bp plastid-like region, the plasmid had no sequence similarities with angiosperm mitogenomes. The plasmid was fully encompassed by Car. avellana or Car. fangiana nuclear sequences from different chromosomes. Two large open reading frames (ORFs), ORF244 (732 bp) and ORF162 (486 bp), were found on the plasmid. BLASTP comparison against the nr database identified homologs of ORF244 in several angiosperm species, including a nearly full-length match in Arabidopsis (AT1G74875, identical 34%). ORF244 homologs were annotated as putative F-box proteins and homologs of ORF162 were annotated as DNA methylation 4 factors in several Rosids. It was unclear whether the two plasmid ORFs were expressed, but there was sufficient evidence to conclude that the plasmid was of nuclear origin.
Genus-specific mitogenome sequences and mosaic origins
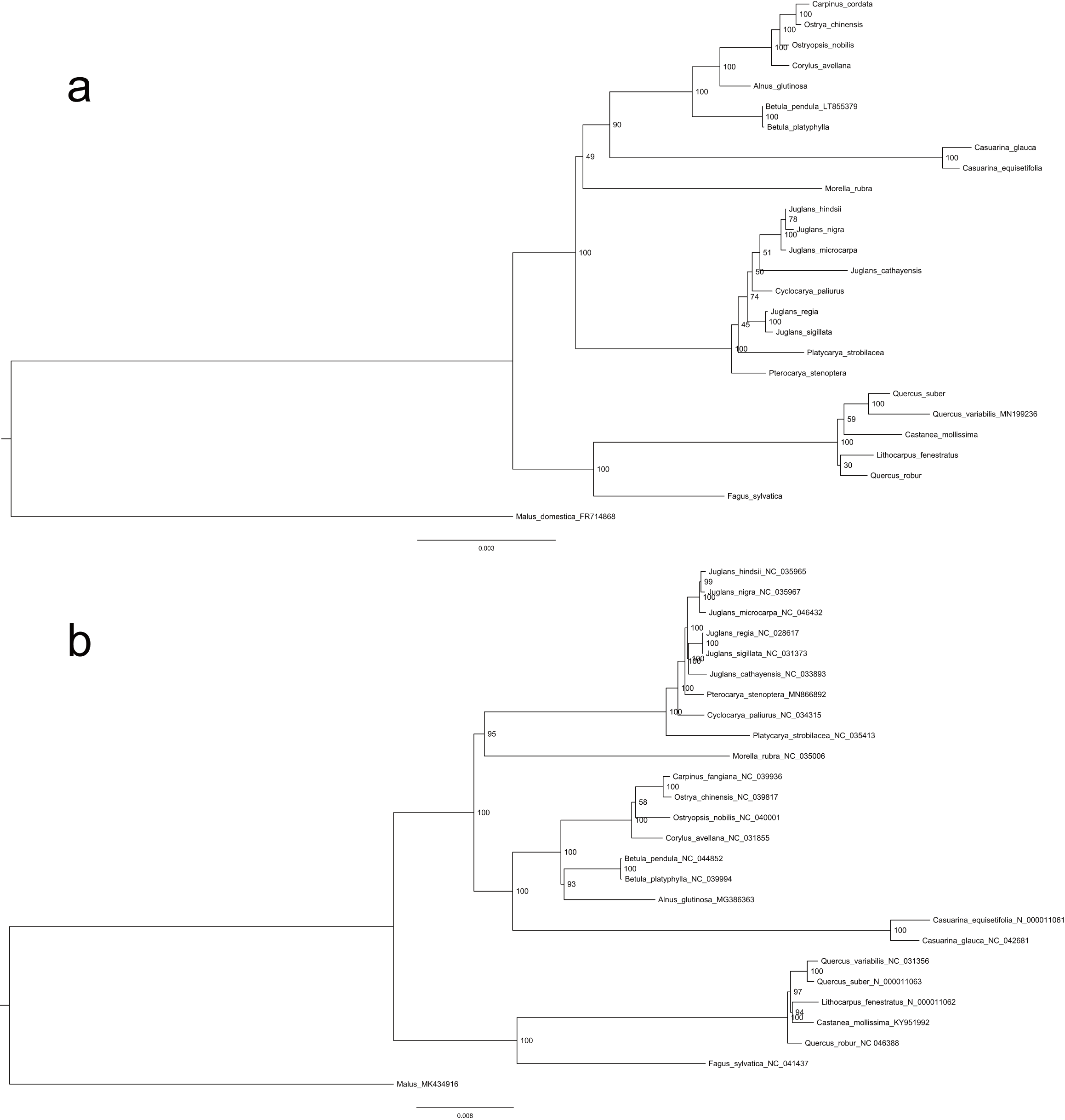
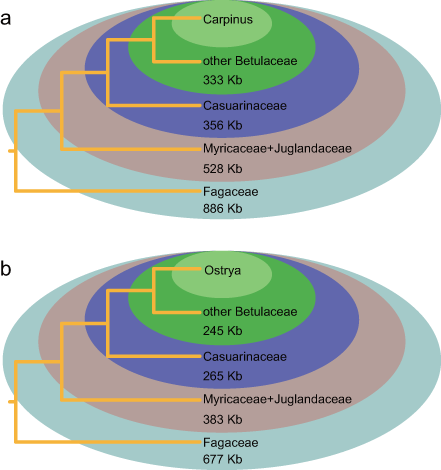
Repeat and MTPT sequences were not solely sufficient to explain the substantial size variation observed among mitogenomes from different species (Table 1). Genus-specific sequences were identified (i.e., sequences with no homologs in other Fagales genera) and used to explore the causes of metagenome size divergence. Quercus species were found to have non-monophyletic relationships (Fig. S7), and Q. robur was not included with other Quercus species when identifying Quercus-specific sequences. Surprisingly, the species with the largest mitogenome, Carpinus, did not contain correspondingly long genus-specific sequences. By contrast, Casuarina species, which had relatively small mitogenomes, had the most unique sequences (Table S5). Plant mitogenomes are prone to absorbing foreign DNA, which might therefore be the source of additional sequences in large mitogenomes. Genus-specific sequences were used to search the NCBI nt database, and best-hits were assessed by order and compartment (Fig. 4; Table S4-S5). Overall, the genus-specific sequences were related to a range of seed plant lineages and were mainly of mitogenomic origin (Fig. 4).
Mitovirus-like sequences were found in several of the 23 Fagales species, including nearly full-length sequences in two Betula species and an approximately 1500 bp sequence in Castanea (Fig. 5). Mitoviruses, which belong to the Narnaviridae family, are positive single-stranded RNA viruses that replicate in host mitochondria. Mitovirus genomes are small, approximately 2.1–4.4 Kb in length, and contain a single ORF encoding a viral RNA-dependent RNA polymerase (RdRP) required for replication (Nibert, 2017). The phylogeny of Fagales mitovirus-like sequences is incongruent with the species tree (Fig. 5), indicating that these sequences were not introduced into Fagales via a single event.
Fagales belong to the nitrogen-fixing lineage of angiosperms, and at least three genera in this study have nitrogen-fixing capacity: Casuarina, Morella, and Alnus (Yelenik and D'Antonio, 2013; Huisman and Geurts, 2020). However, there was no indication that these genera contained more sequences similar to bacteria than other Fagales species.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}