Method overview
To enhance discovery of relevant biomarkers, we extended the commonly used machine learning approach LASSO to incorporate biological information about the features by applying specialized regularization. In a typical dependency biomarker analysis, the dependent variable is the dependency score of the target gene and the independent variable is a genome-wide molecular profile, such as CN variation (Fig. 1A). Sparsity promoting regularization techniques, such as LASSO, are a popular choice for biomarker discovery because these methods aim to identify a small set of highly informative features in high dimensional data such as molecular profiles (Hédou et al. 2024). Therefore, we adapted the following two-step procedure to the LASSO regularization.
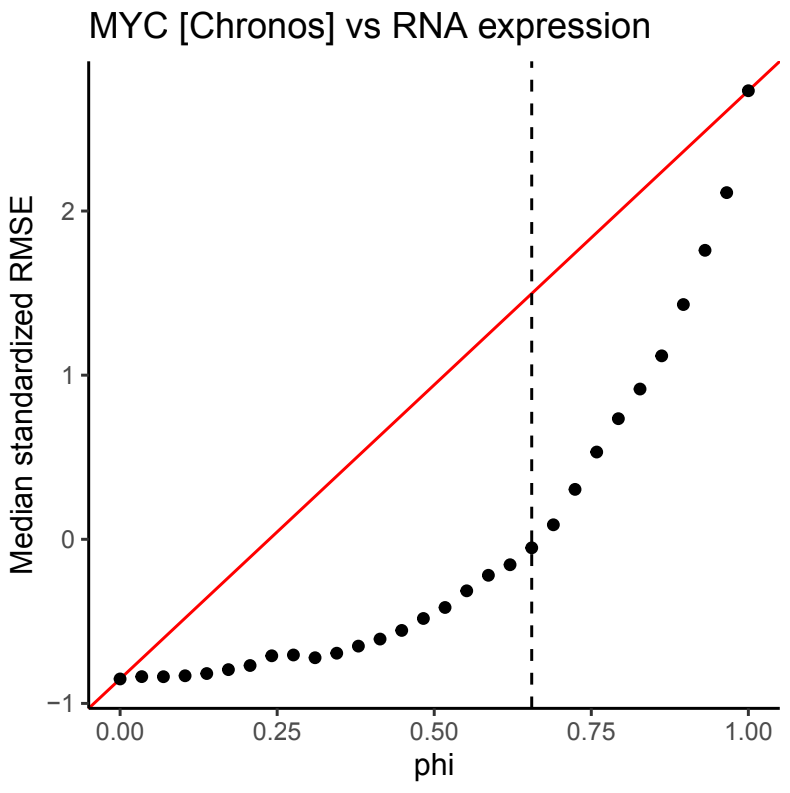
In the standard LASSO model, the regularization parameter \(\:\lambda\:\) is optimized using cross-validation (Fig. 1B). The \(\:\lambda\:\) parameter defines the amount of shrinkage each input feature receives. Optimization of this parameter concludes the regularization of the traditional LASSO, and we term the resulting model the baseline LASSO model. Upon optimization of \(\:\lambda\:\), we introduce a novel parameter Φ which represents the magnitude of prior evidence linking each feature to the target gene. This evidence may be derived from PPI databases such as STRING DB but is not limited to this data. The Φ parameter is optimized following analogous cross-validation procedure. We term the resulting model using the optimized \(\:\lambda\:\) and Φ parameters the bio-primed LASSO model. For selecting informative biomarkers, the feature coefficients of each model were assessed and interpreted.
Predict MYC dependency using RNA expression biomarkers
We first applied our method to the Chronos dependency data set derived from genome-wide CRISPR knockout experiments for 17,386 genes across 1,048 cancer cell lines (Dempster et al. 2021). We set out to find RNA expression biomarkers to predict the dependency of oncogene c-Myc (MYC). RNA expression data was filtered to 12,182 expressed genes and subsequently z-score normalized. This set of genes was used as input features to discover relevant biomarkers to predict MYC dependency.
Using 10-fold cross validation, a value of 0.65 was inferred for the Φ parameter (Fig S1). A total of 188 features were assigned non-zero coefficients in the bio-primed model and deemed as relevant biomarkers (Fig. 2A). The largest coefficient, which represents the most informative feature, was assigned to RNA expression of the MYC gene itself. Both the baseline and bio-primed models identified MYC RNA expression as the major predictor, consistent with the paradigm of oncogene addiction (Weinstein 2002). Additionally, significant correlation was observed between the coefficients derived from the two models for the remaining predictors (Fig. 2B). We next calculated the correlation between each input feature and the target dependency. Overlaying this information on top of the coefficients derived from the two models revealed that predictors with positive and negative LASSO coefficients also showed positive or negative correlation, respectively (Fig. 2C). As expected for an oncogene, MYC RNA expression levels were negatively correlated with MYC dependency. Cell lines with elevated RNA expression of MYC were more dependent on MYC (Fig. 2D).
Of note, a fraction of RNA biomarkers including STAT5A and NCBP2 received non-zero coefficients exclusively in the bio-primed and not the baseline model (Fig. 2C). STAT5A, a member of the Signal Transducer and Activator of Transcription (STAT) family, has previously been identified as a potent inducer of MYC (Villarino et al. 2022; Preston et al. 2015; Lord et al. 2000). As an inducer of MYC, the oncogene addiction model suggests that elevated STAT5 should be a biomarker in MYC-driven cancers. Correspondingly, we observed increased MYC dependency in cell lines with high expression of STAT5 RNA (Fig. 2D).
NCBP2 (also known as Nuclear Cap-Binding Protein Subunit 2) is a component of the cap binding complex and is required for the recruitment of splicing machinery to nascent mRNA (Görnemann et al. 2005; Mazza et al. 2001). We, and others, have previously shown that MYC driven cancers are vulnerable to perturbation of the spliceosome (Hsu et al. 2015; Koh et al. 2015). We observed that cell lines with low levels of NCBP2 RNA were more dependent on MYC (Fig. 2D). The observed correlation between the RNA levels of STAT5A and NCBP2 with MYC dependency implies these genes as relevant biomarkers of MYC dependency.
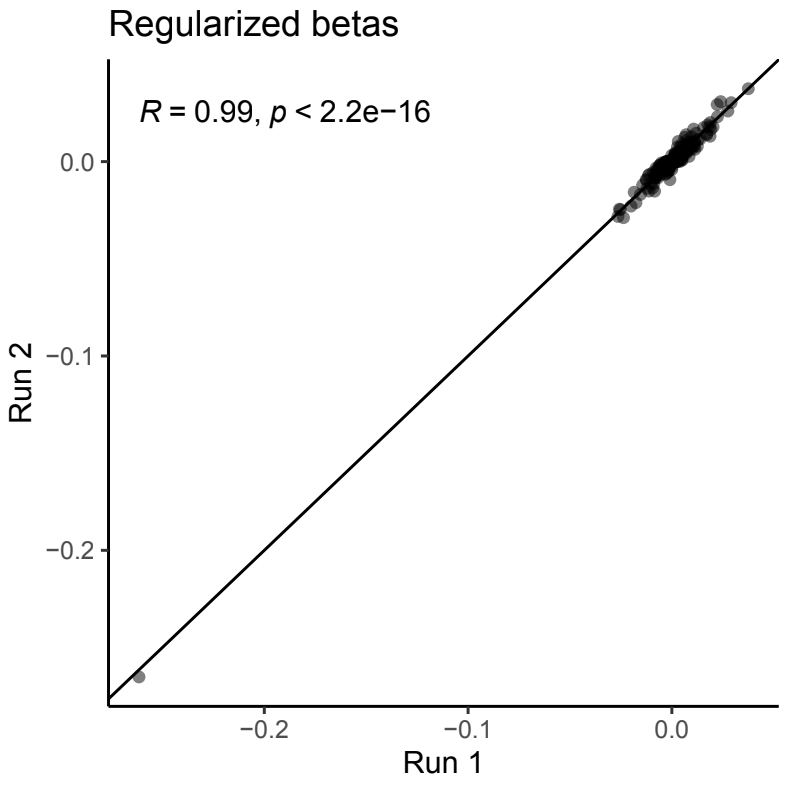
To assess the computational robustness of our method, we performed a second independent run using the same input features and MYC dependency as the outcome variable. The bio-primed model’s coefficients derived from these two independent runs showed strong correlation, demonstrating high reproducibility across runs (Fig S2).
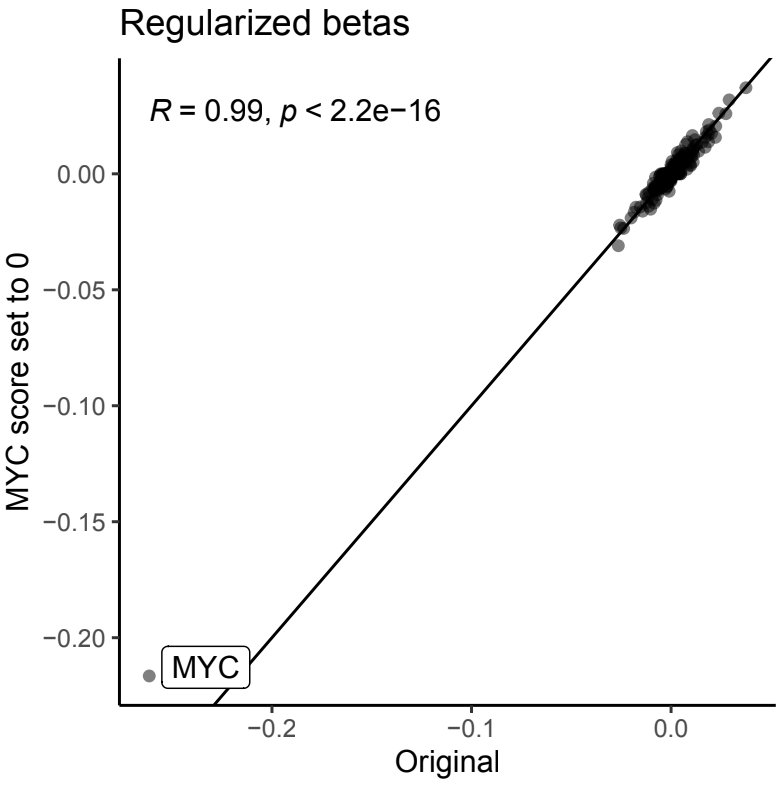
To further assess the robustness of our model to noise in the provided biological network information, we manually set the evidence score for MYC to 0, the minimum evidence score value. In this way, the model will not be encouraged to favor MYC RNA expression as one of the biomarkers. Nonetheless, our model assigned a large coefficient to MYC RNA expression, demonstrating that our model facilitates discovery of novel associations without prior supporting biological data and is robust to incomplete network annotations (Fig S3).
Predict EGFR dependency using copy number biomarkers
We next applied our method to the Demeter2 (D2) dependency data set. This gene dependency data was derived from genome-wide short hairpin RNA screen experiments for 17,309 genes across 707 cancer cell lines (Tsherniak et al. 2017). As a second use case, we set out to discover CN biomarkers predicting EGFR dependency as measured using the D2 score. Linkage disequilibrium (LD) in CN profiles makes it particularly difficult to extract relevant biomarkers since many genes will carry comparable statistical evidence of association.
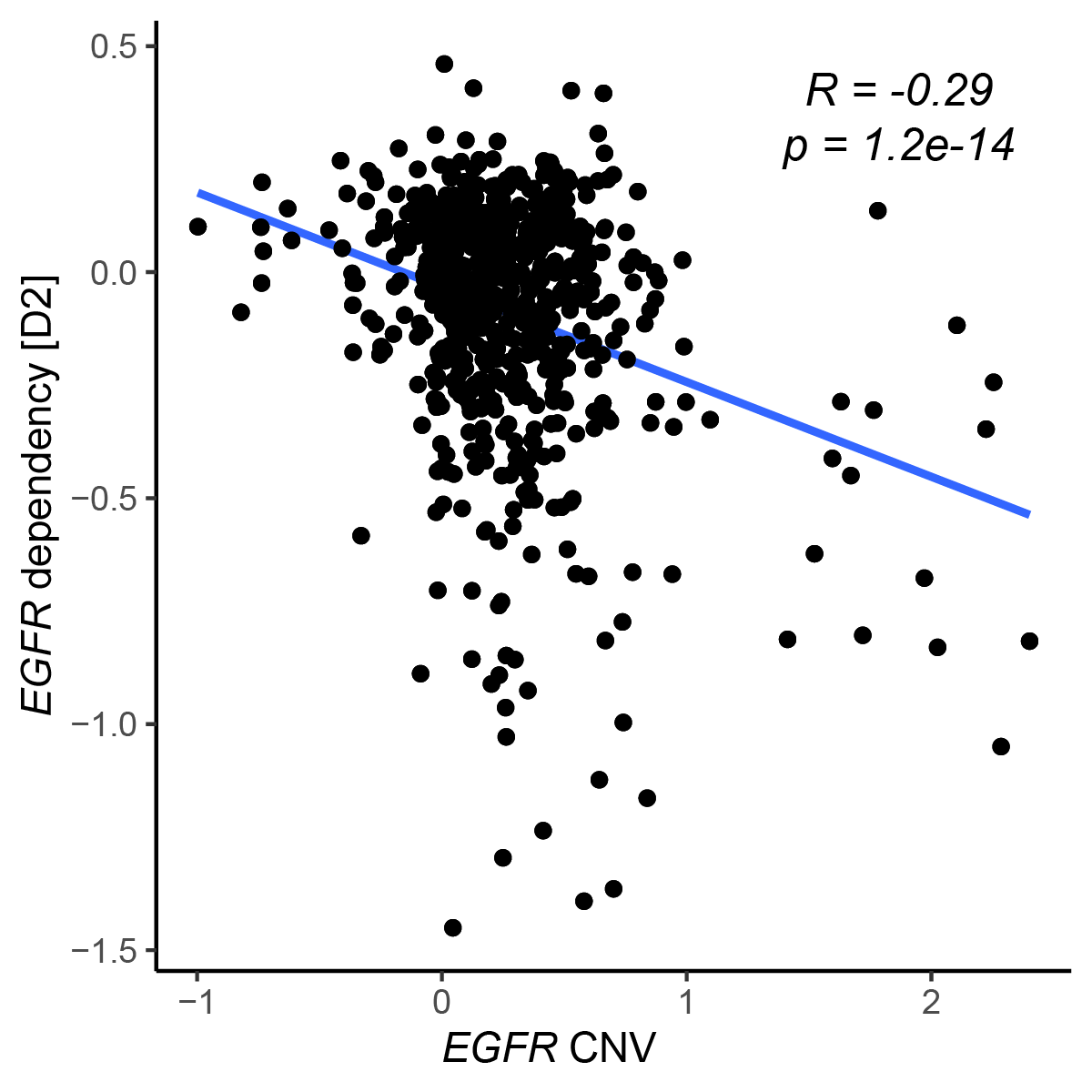
We first calculated the correlation coefficient between EGFR dependency and the CN estimate of each gene. Visualization of this genome-wide correlation profile revealed a strong negative correlation between EGFR CN and EGFR dependency (Fig. 3A). As expected from an oncogene, amplification of EGFR CN conferred EGFR dependence on the cells (Fig S4).
Interestingly, we observed a second peak with moderate negative correlation on chromosome 11 (highlighted in a purple box). Focusing on this locus revealed many genes with strong negative correlation between CN and EGFR dependency (Fig. 3B). The strong LD structure makes it difficult to select a specific biomarker from this region based on correlation coefficients alone. The baseline model assigned a single non-zero coefficient to the USP35 gene in this region. To the best of our knowledge, there exists no reported connection between USP35 and EGFR. STRING also did not assign an association score for these two genes. We believe that the reason the baseline model picked the USP35 gene is due to a spurious association with the underlying driver gene. The bio-primed model, on the other hand, identified GAB2 CN as the most informative biomarker in this region based on the magnitude of the LASSO coefficient (Fig. 3B). GAB2 (GRB2-associated binding protein 2) is an adaptor protein that plays a critical role in transmitting signals from receptor tyrosine kinases, such as EGFR, to downstream pathways involved in cell proliferation, survival, and migration (Adams, Aydin, and Celebi 2012). Amplification of GAB2 can lead to increased activation of the PI3K/AKT and MAPK pathways, both of which are downstream effectors of EGFR signaling (Gu et al. 1998), supporting the idea that GAB2 amplification may potentiate oncogenic processes driven by EGFR and increase the dependency of cancer cells on EGFR activity. Indeed, stratifying the cell lines by EGFR and GAB2 CN gain revealed that simultaneous gain of EGFR and GAB2 CN significantly increased dependency on EGFR, linking the GAB2 CN to EGFR dependency (Fig. 3C).
These data suggest that patients with GAB2 amplification may be more sensitive to drugs targeting EGFR. To explore this hypothesis, we analyzed existing drug sensitivity data provided by the DepMap resource. We correlated drug sensitivity profiles for 545 drugs from the Cancer Target Discovery and Development Network with GAB2 CN profiles. Several of the most strongly associated drug sensitivities were indeed EGFR inhibitors (Fig. 3D). For example, cell lines with GAB2 amplification but CN neutral EGFR showed increased sensitivity to EGFR inhibitor Afatinib (Fig. 3E).
To systematically evaluate our method, we used the D2 dependency data. We first identified a total of 453 highly selective genes that showed strong dependency in a subset of cell lines. Given their selective dependency profile these genes represent promising cancer drug targets. We hypothesized that the selective dependency profile may be driven by CN aberrations of the gene itself of biologically relevant genes. Therefore, each of these gene dependencies was subjected to CN biomarker analysis using the baseline and bio-primed model approaches, with the goal of identifying the underlying genomic aberrations driving the dependencies. Out of 453 target genes, 432 identified at least one predictive CN biomarker in either approach (Table S1).
Next, we set out to compare the biomarkers derived from the baseline and bio-primed models. For each gene and to ensure discriminative power, two sets of mutually exclusive biomarkers were defined: 1) The top 20 biomarkers with a positive coefficient derived from the bio-primed model and not identified using the baseline model. 2) The top 20 biomarkers with a positive coefficient derived from the baseline model and not identified using the bio-primed model. For each of these gene sets, we calculated the co-dependency between each target and the corresponding biomarkers using Pearson correlation.
For example, UTP4, a key component of the processome, a large ribonucleoprotein complex involved in the early steps of ribosome biogenesis (Freed et al. 2012), showed multiple peaks of correlations between CN and dependency (Fig. 4A). The bio-primed model identified UTP4 in the peak with the strongest genome-wide association on chromosome 16. This result was consistent with the so-called CYCLOPS model, which posits that partial loss of CN yields cancer specific liabilities (Paolella et al. 2017). The baseline approach failed to identify UTP4 and instead selected unrelated biomarkers near the UTP4 locus. To the best of our knowledge, none of these markers have previously been linked to UTP4 biology.
We generalized this approach across the remaining 431 genes. This comprehensive analysis demonstrated that biomarkers derived from the bio-primed model showed significantly stronger co-dependency with the target compared to biomarkers derived from the baseline model (Fig. 4D). Thus, the biomarkers identified by the bio-primed approach are more relevant to the target biology compared to the baseline approach. A full list of results may be found in Table S1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}