RGS12 is expressed in placental cell mitochondria, cytoplasm, and nucleus
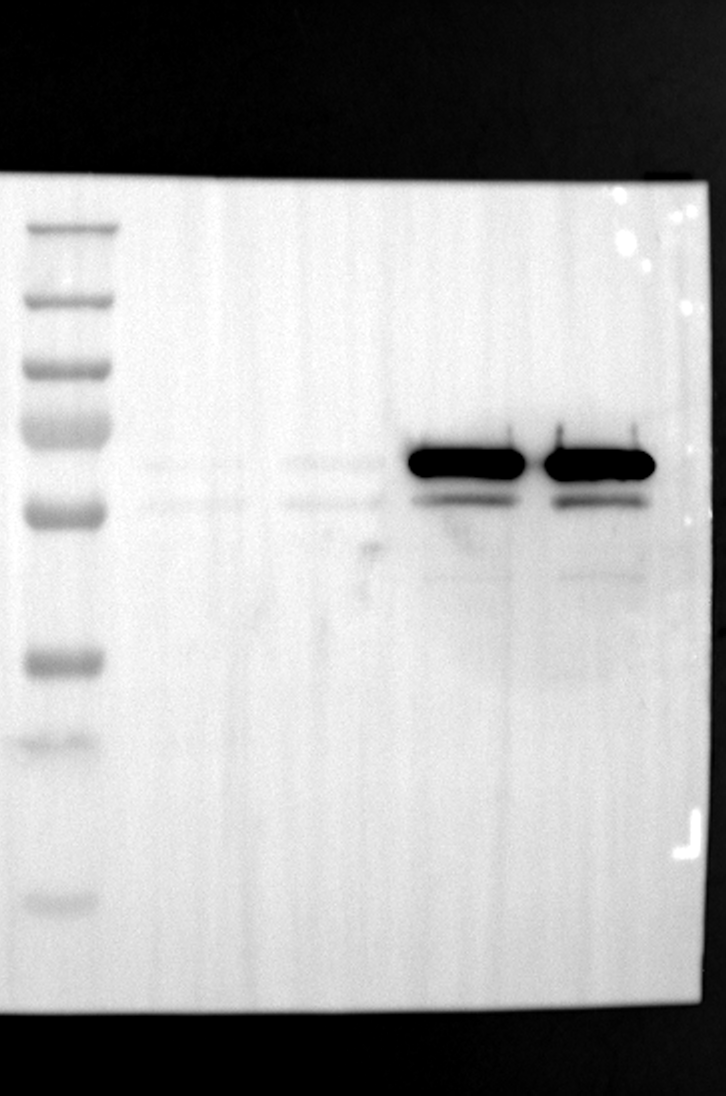
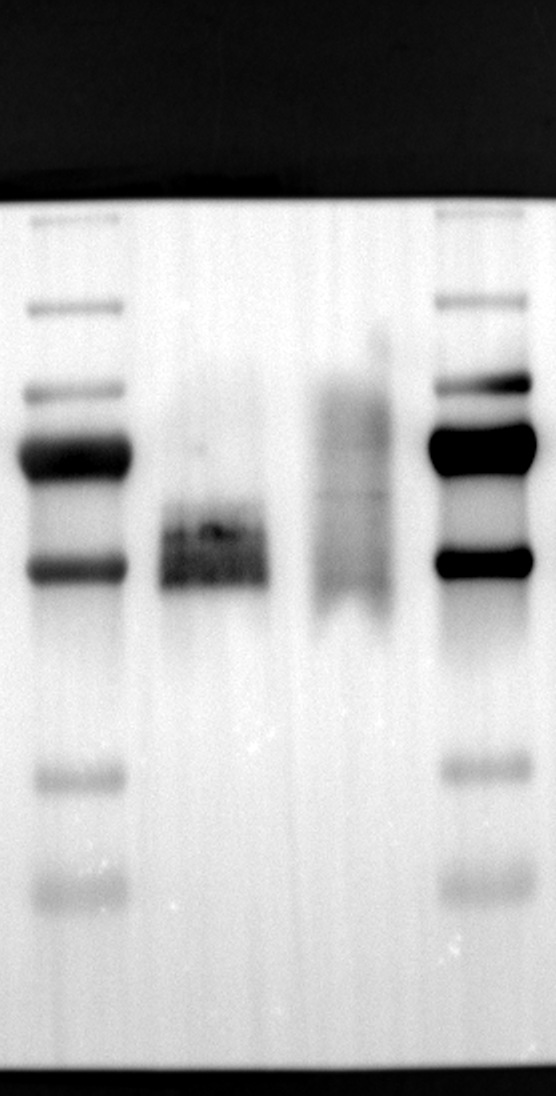
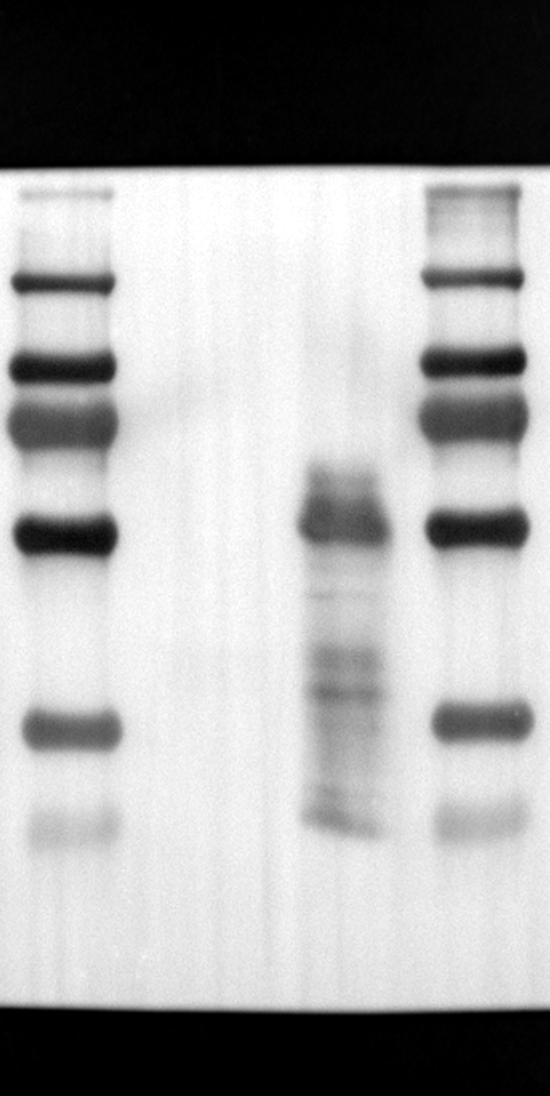
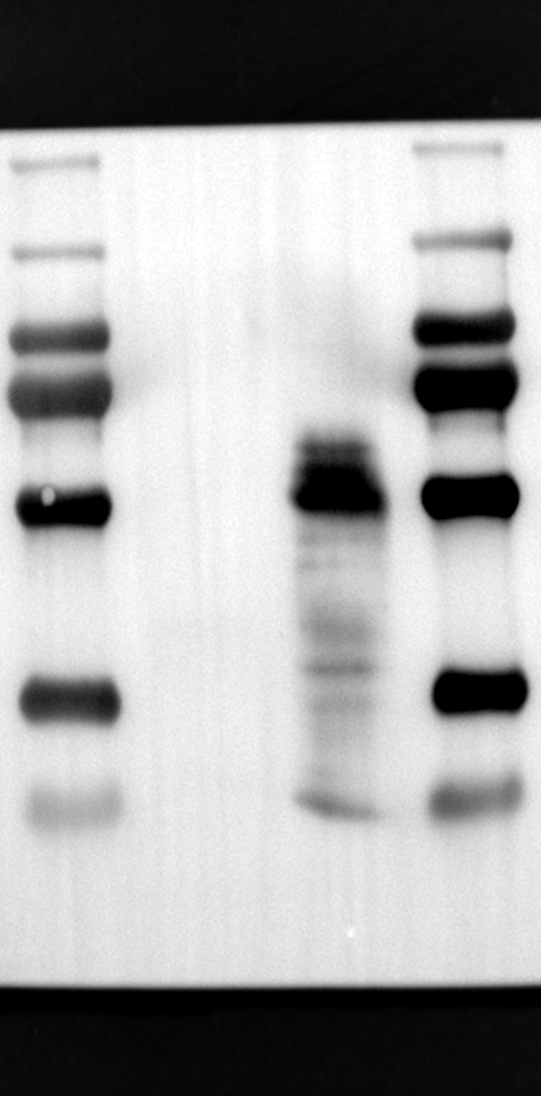
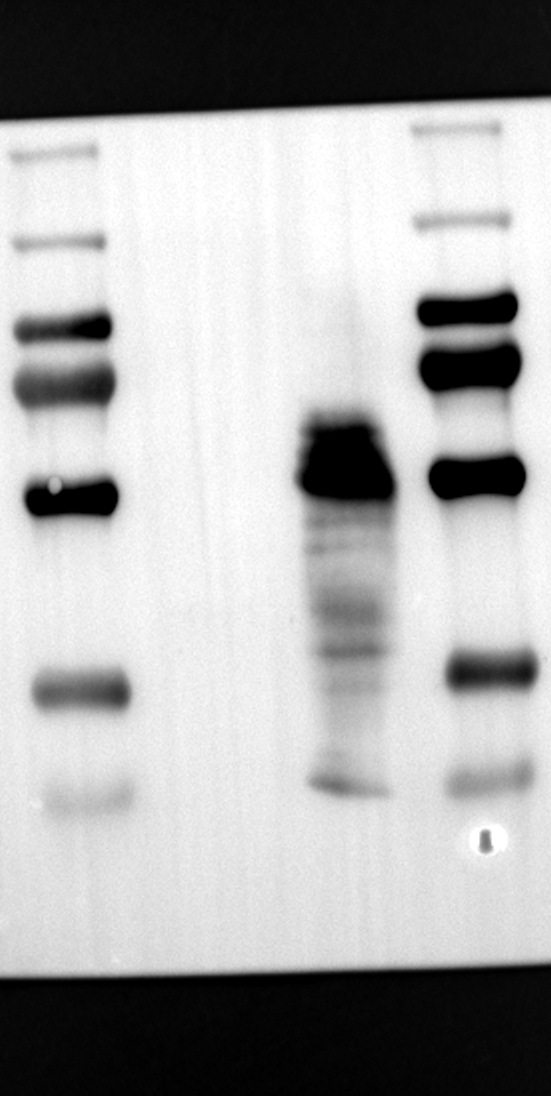
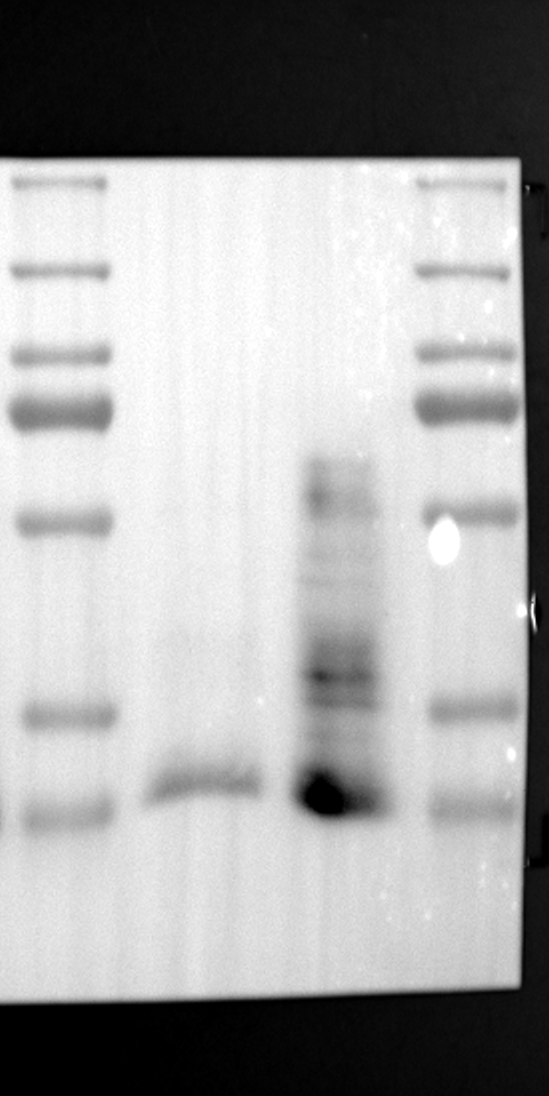
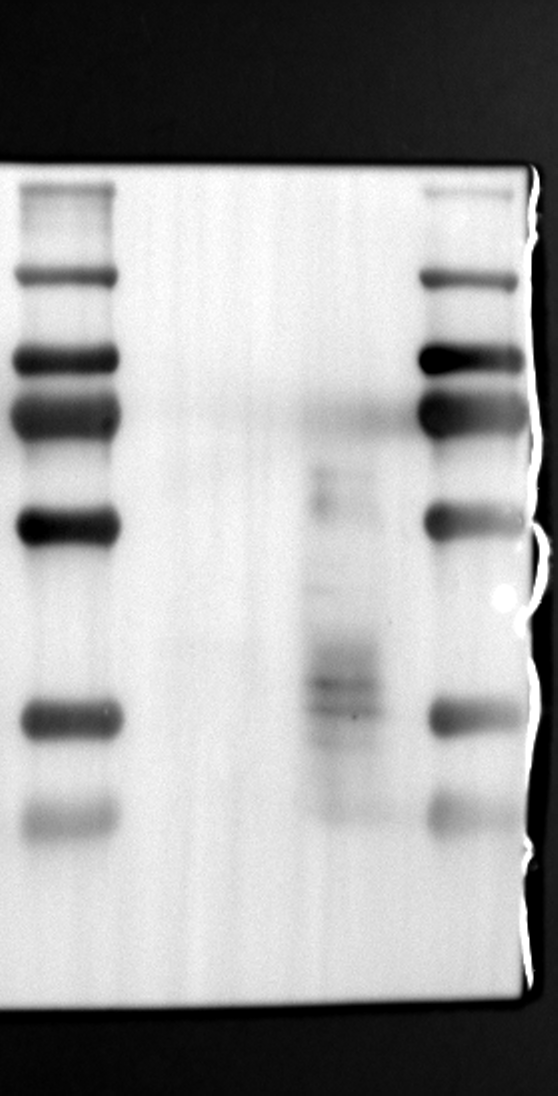
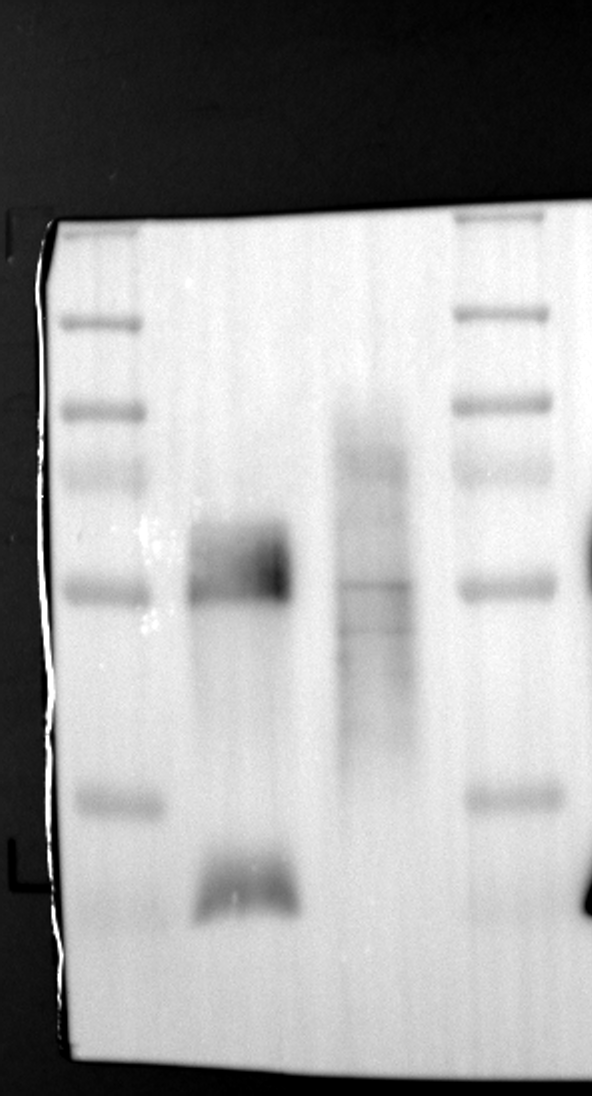
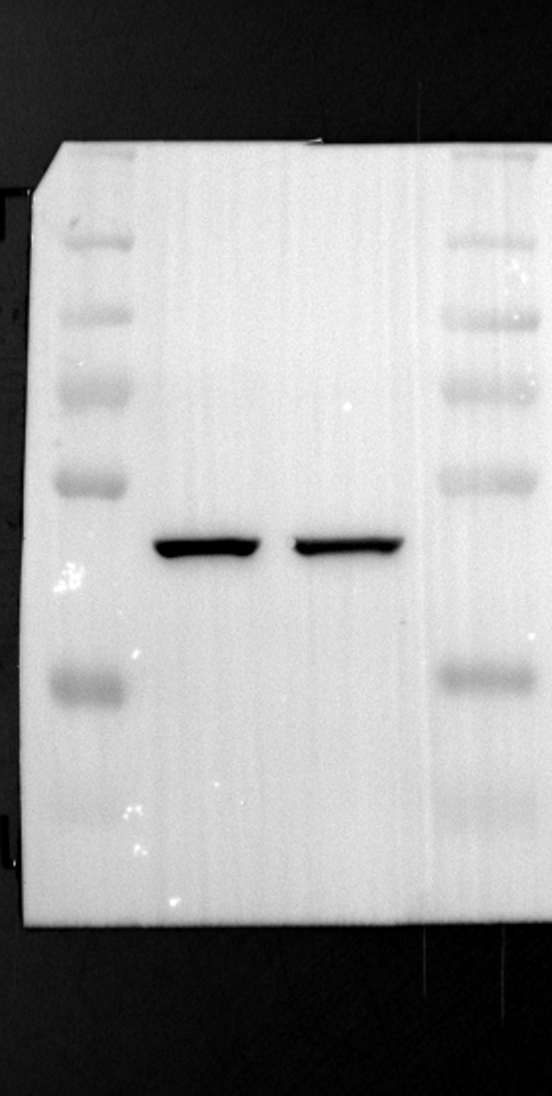
RGS12 has been implicated in various cellular processes, including proliferation, differentiation, oxidative phosphorylation, ROS production, and apoptosis, suggesting its potential role as a key regulator within mitochondria [34, 35]. To investigate its localization in placental cells, we performed immunostaining on the human placental trophoblast cell line HTR-8/SVneo. Nuclei were stained with DAPI (Fig. 1A), RGS12 was visualized with an anti-RGS12 antibody (Fig. 1B), and mitochondria were labeled with MitoTracker (Fig. 1C). Co-localization analysis revealed significant overlap between RGS12 and the mitochondrial marker, suggesting a primarily mitochondrial localization for RGS12 in these cells. Partial co-localization with the nuclear marker DAPI was also observed (Fig. 1D-F). To further characterize RGS12 localization and expression in placental tissues, we examined protein extracts from both mouse (18.5 days post-coitum, dpc) and human (term birth, TB) placentas. Mitochondrial and cytoplasmic fractions were isolated, and immunoblotting was performed to detect RGS12 alongside α-tubulin (cytoplasmic marker) and HSP60 (mitochondrial marker). RGS12 was detected in both mitochondrial and cytoplasmic fractions of both mouse and human placentas (Fig. 1G and H), with a significantly higher abundance in the mitochondrial fractions (approximately 5–10 fold), which may be attributed to some extent to other cell types within the placental tissue. To verify the mitochondrial enrichment of RGS12, specifically in trophoblast cells, we performed immunoblotting on mitochondrial and cytoplasmic extracts from human placental trophoblast cell lines HTR-8/SVneo and Swan71. Interestingly, RGS12 displayed comparable expression levels in both the mitochondrial and cytoplasmic fractions of these cell lines (Fig. 1I and J). Furthermore, its overall expression was correlated with the mitochondrial content of the cells. These findings suggest that while RGS12 is present in both cellular compartments, its expression may be linked to the abundance of mitochondria within placental trophoblast cells.
RGS12 regulates the tyrosine phosphorylation of mitochondrial ATP5B in placental trophoblast cells
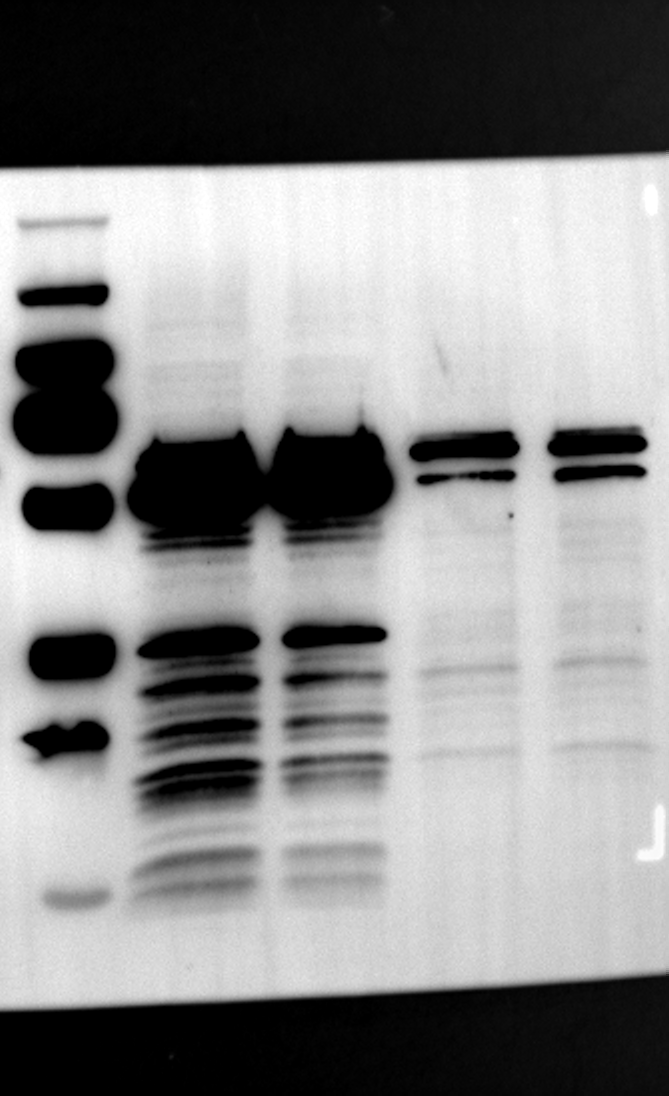
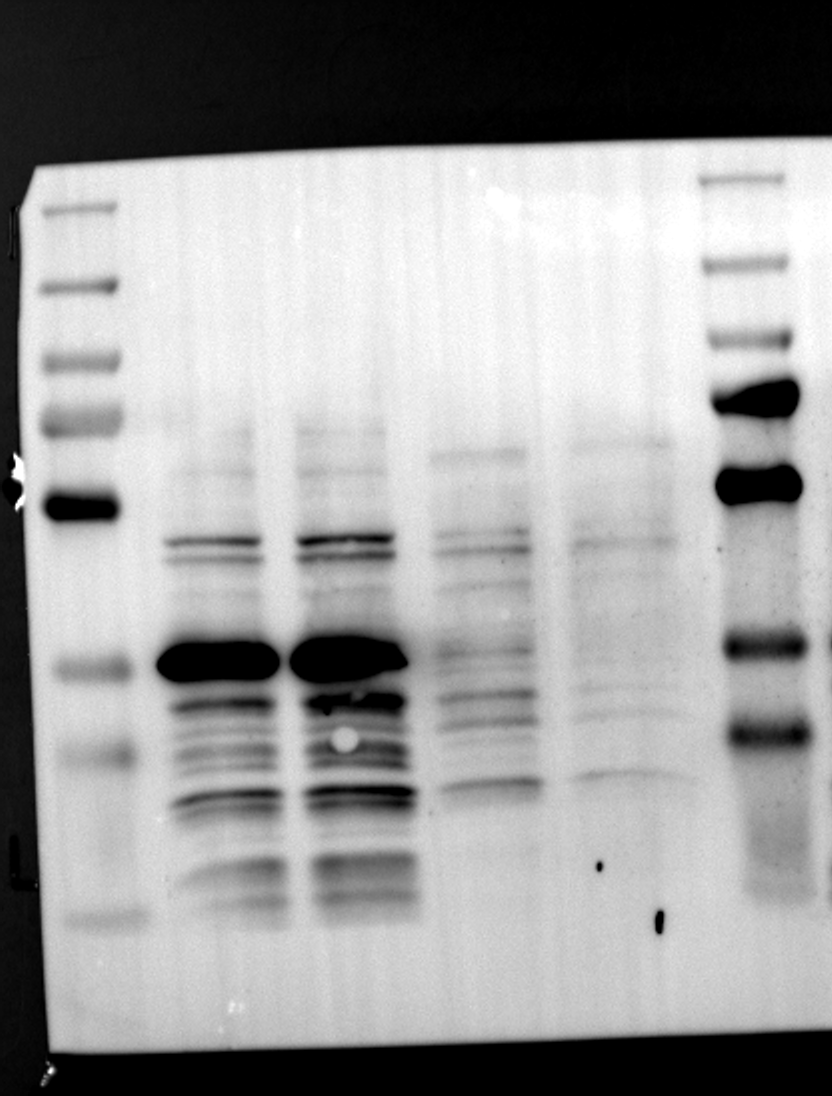
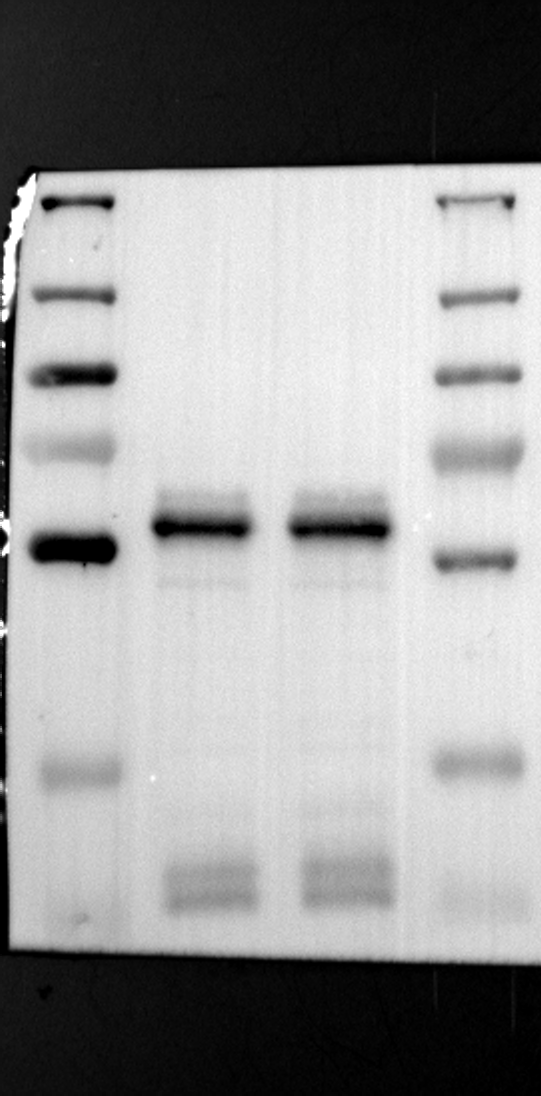
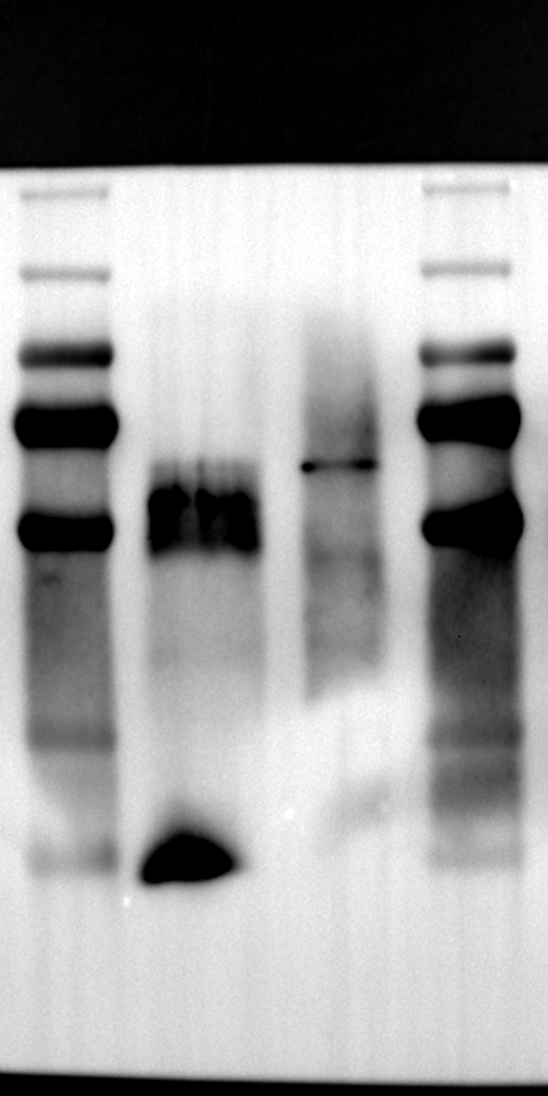
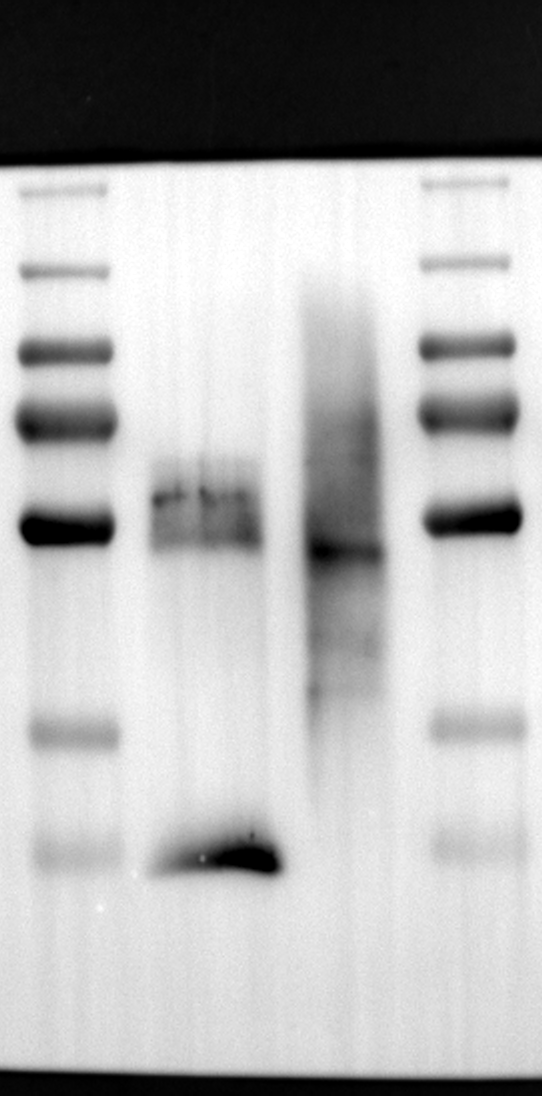
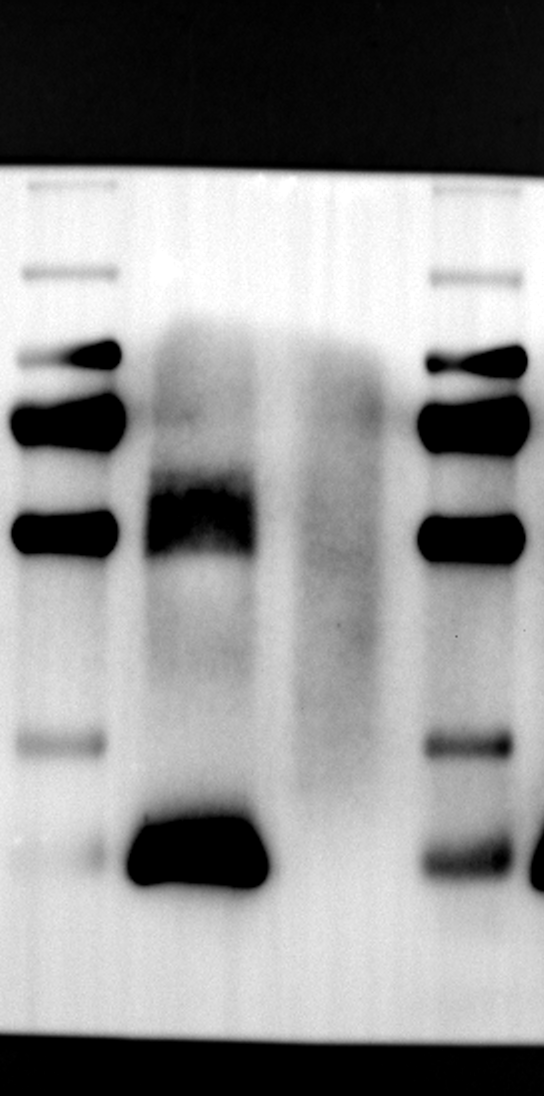
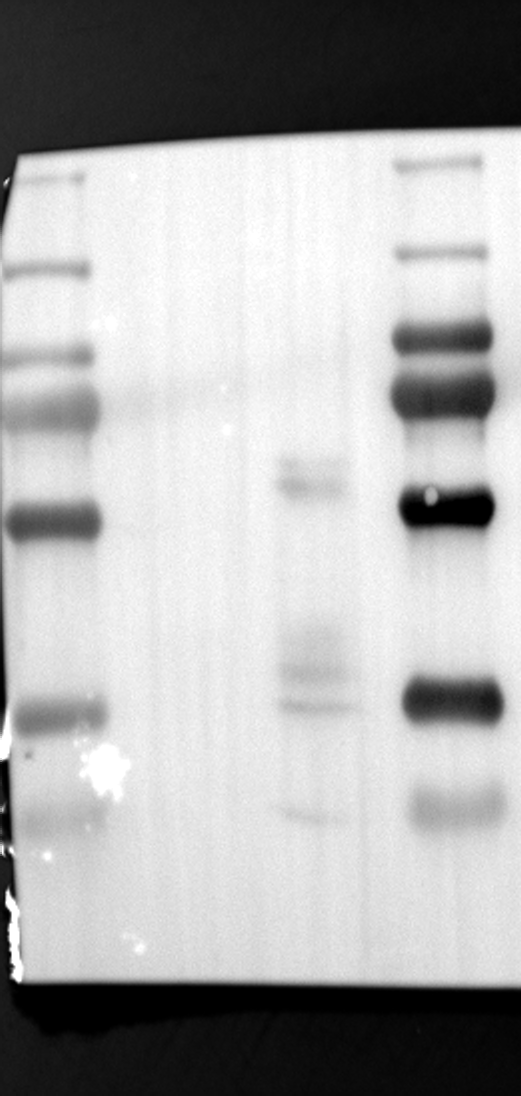
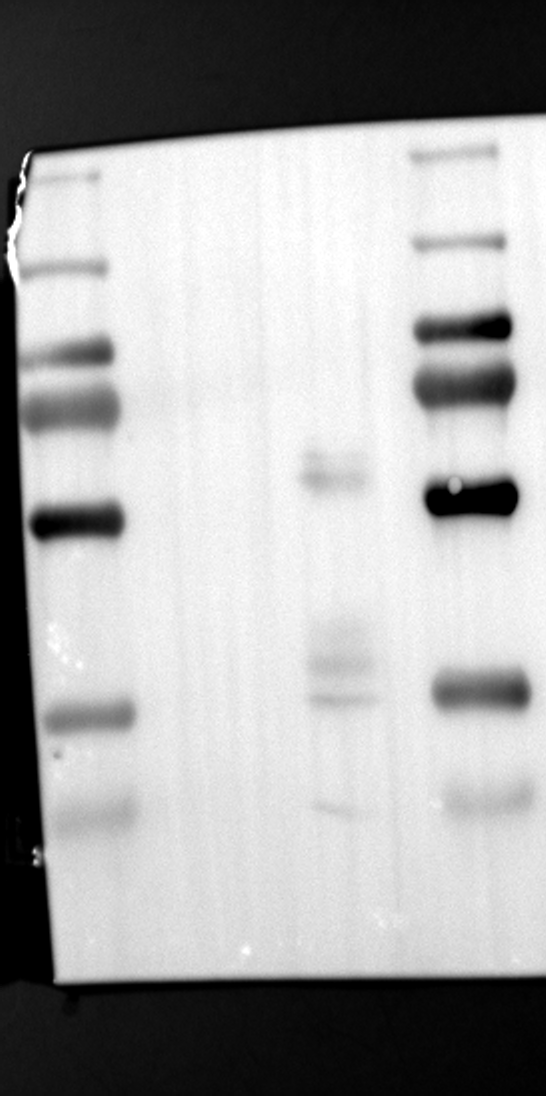
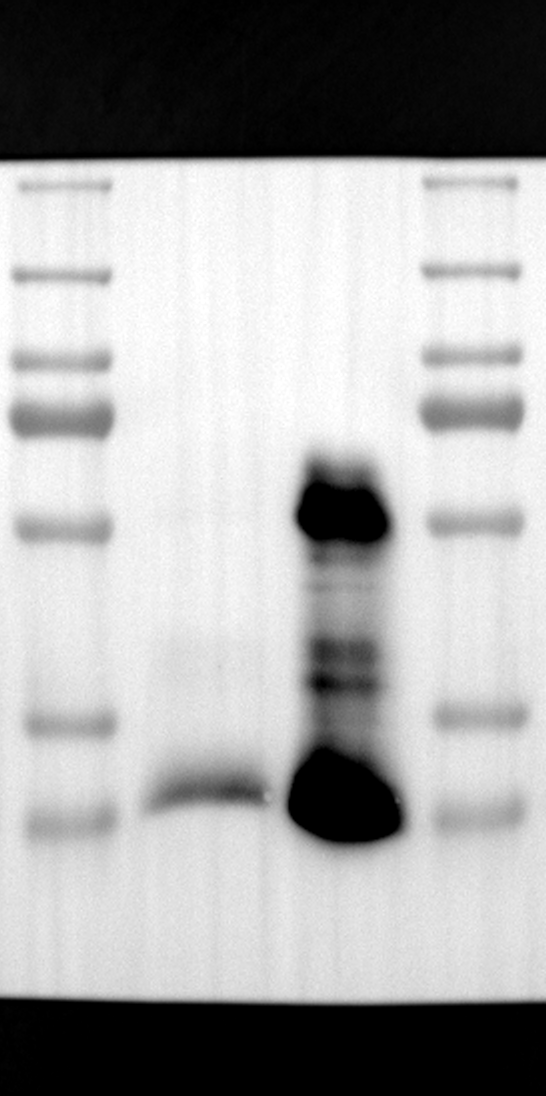
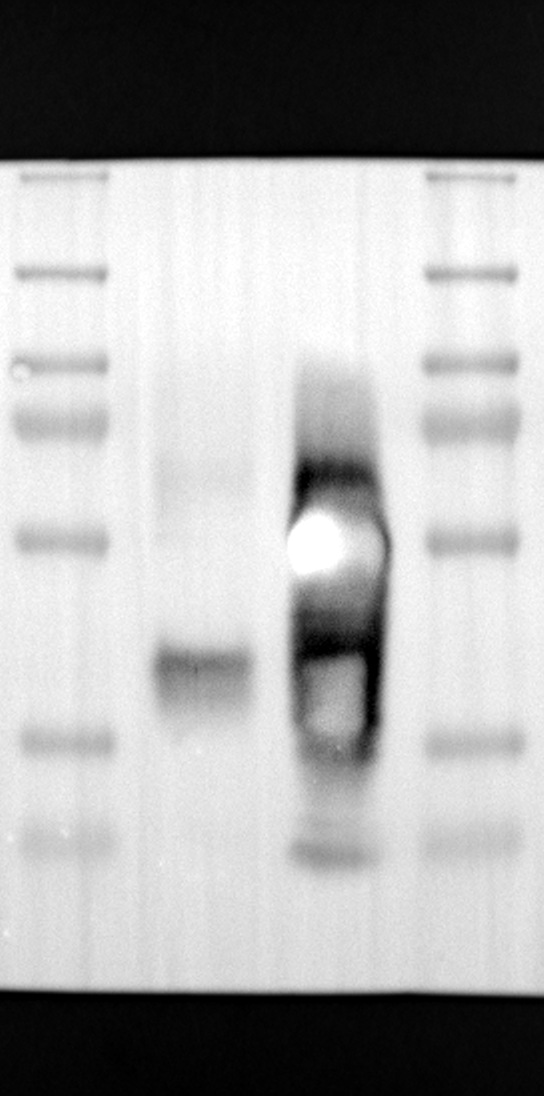
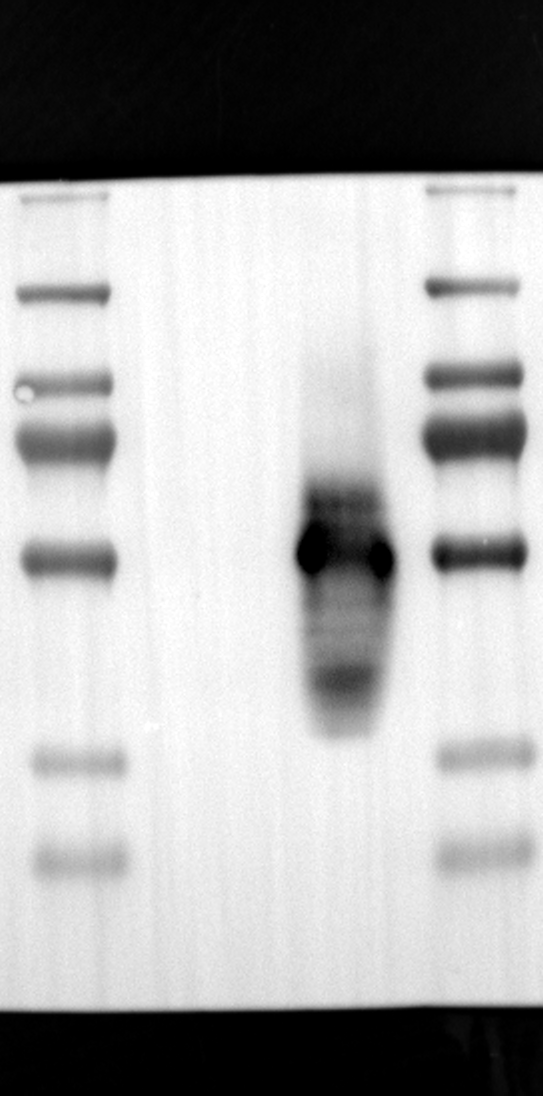
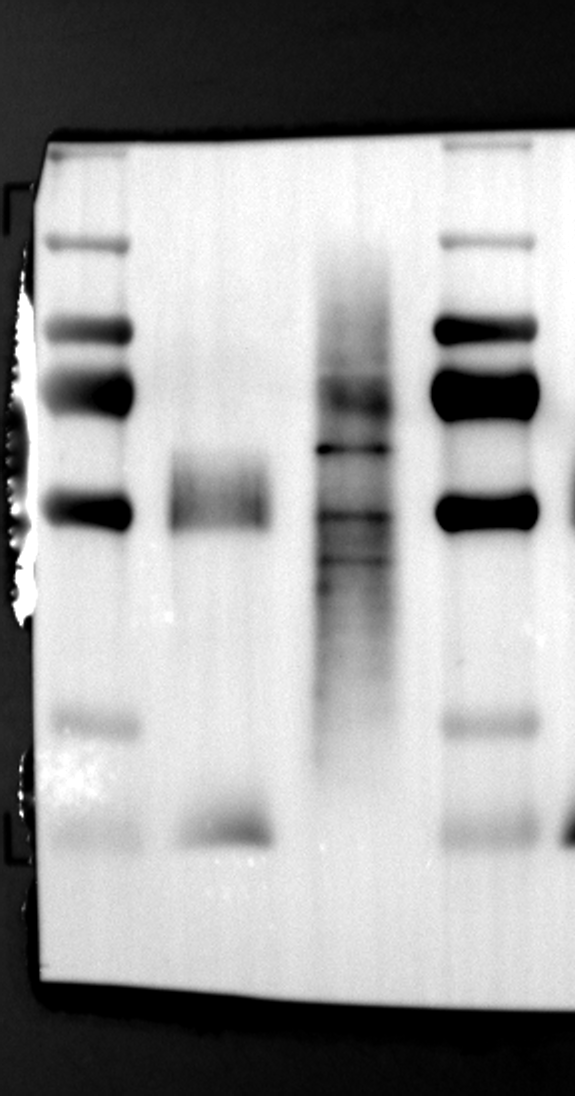
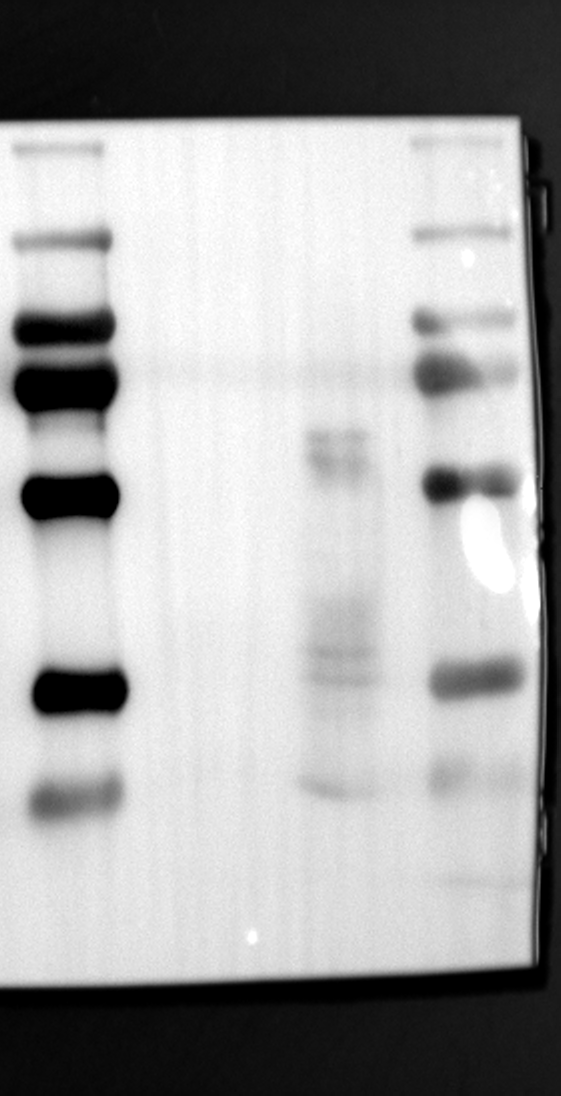
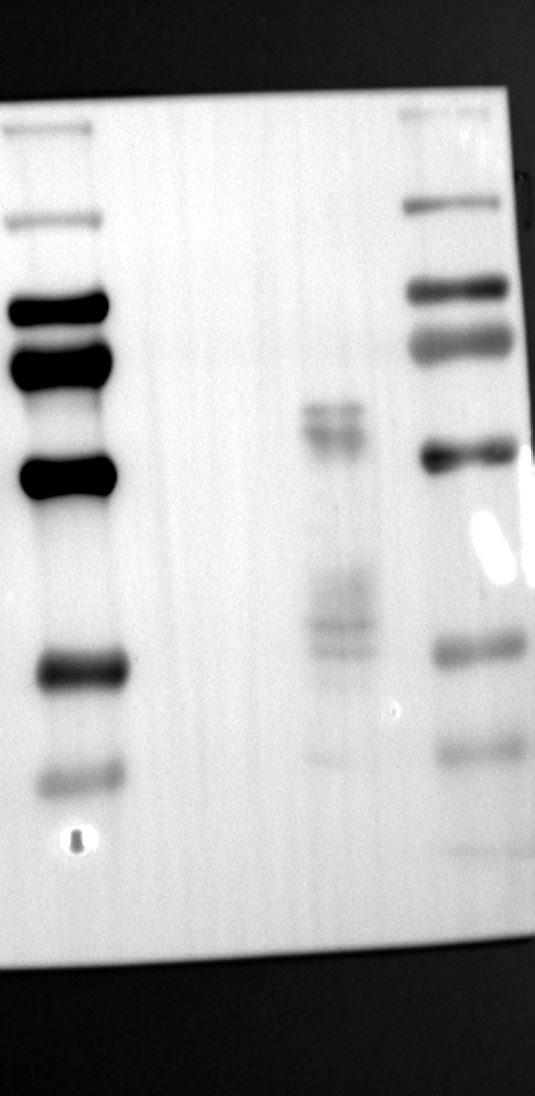
Having identified RGS12 localization within placental trophoblast cells, we next explored its potential mechanisms of action. Immunoprecipitation (IP) experiments using RGS12 antibodies followed by mass spectrometry analysis revealed high-affinity binding between RGS12 and ATP5B, with minimal interactions detected between RGS12 and other mitochondrial proteins (Table S1). This finding suggests a specific role for RGS12 in mediating mitochondrial functions through ATP5B in these cells. To validate this interaction, we performed co-IP experiments using specific antibodies against ATP5B and RGS12 in placental trophoblast cell lines (HTR-8/SVneo and BeWo) and placental tissues (mouse 18.5 dpc and human TB). As expected, both proteins co-immunoprecipitated with their respective antibodies (Fig. 2A-G), supporting the hypothesis that ATP5B may be a regulatory target of RGS12 in placental trophoblast cells. Further confirmation of their colocalization was obtained through immunofluorescence (IF) staining of mouse and human placental tissues (Fig. 2H and I). Considering that RGS12 regulates various cellular processes through the phosphorylation of target proteins and that tyrosine phosphorylation of ATP synthase subunits is critical for its function [36–38], We investigated the potential role of RGS12 in phosphorylated tyrosine (p-Tyr) modification of ATP5B. Co-IP experiments using p-Tyr and ATP5B antibodies revealed phosphorylation of ATP5B in all tested samples (placental trophoblast cell lines, mouse, and human placentas) (Fig. 3A-G). These findings suggest a possible role for RGS12 in regulating ATP5B p-Tyr status. To functionally test this hypothesis, we manipulated RGS12 expression in HTR-8/SVneo cells. RGS12 knockdown (ShRGS12) and overexpression (RGS12OE) were achieved (Fig. S1A-D). Interestingly, RGS12 knockdown specifically decreased mitochondrial p-Tyr levels without affecting overall cytoplasmic phosphorylation (Fig. 3H and I), indicating a specific role for RGS12 in regulating mitochondrial protein phosphorylation. Furthermore, RGS12 knockdown specifically reduced ATP5B mRNA and protein expression levels without affecting ATP5A (Fig. S1E-H). Collectively, these data suggest that RGS12 may regulate mitochondrial function in placental trophoblast cells by modulating the expression and tyrosine phosphorylation of ATP5B.
Knockdown of RGS12 in HTR-8/SVneo cells leads to mitochondrial dysfunction
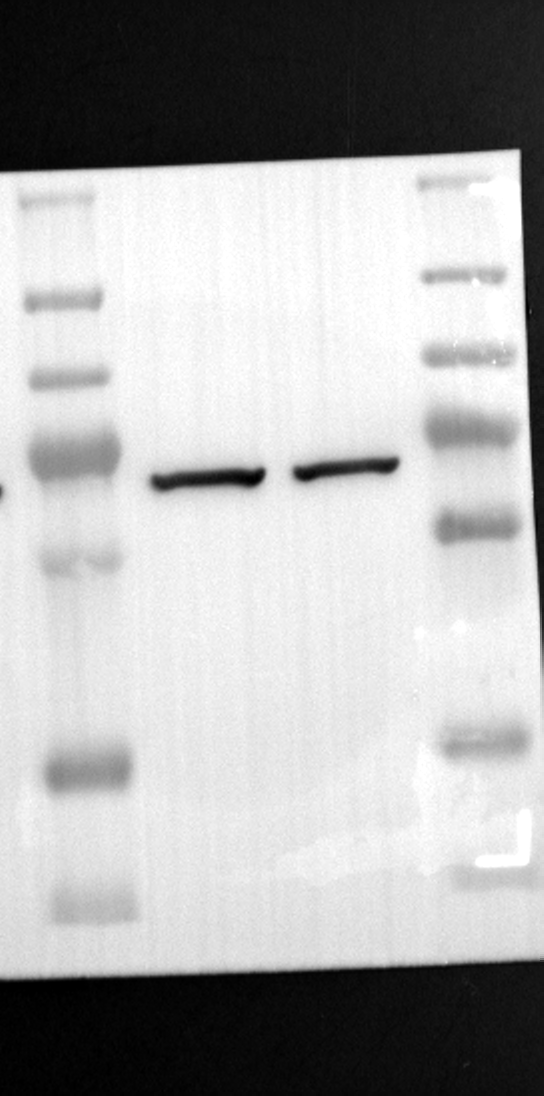
To investigate the functional consequences of RGS12 knockdown on mitochondrial function in placental trophoblast cells, we established stable cell lines. HTR-8/SVneo cells were transfected with either scramble control shRNA (shCTL) or shRNA targeting RGS12 (shRGS12). Western blot analysis confirmed a significant reduction in RGS12 protein levels in both the cytoplasm and mitochondria of shRGS12 cells compared to the control group, with a more pronounced decrease observed in the mitochondrial fraction (Fig. 4A). Transmission electron microscopy (TEM) was employed to evaluate mitochondrial morphology. Compared to the control group, shRGS12 cells displayed significant alterations in mitochondrial morphology. These included swelling, rounding, a dissolved intramembranous matrix, reduced diameter and area, and a notable increase in damaged mitochondria (Fig. 4B-D). As anticipated, shRGS12 knockdown resulted in a significant decrease in both mitochondrial DNA (mtDNA) copy number and cellular ATP content in HTR-8/SVneo cells compared to the control group (Fig. 4E and F). To further assess mitochondrial function, we utilized MitoTracker for mitochondrial staining and Dihydroethidium staining to detect basal mitochondrial ROS production (Fig. 4G). These experiments revealed a reduction in mitochondrial abundance and a concomitant increase in ROS production in shRGS12 cells compared to the control group (Fig. 4G and H). Finally, we examined the impact of RGS12 knockdown on mitochondrial antioxidant function in HTR-8/SVneo cells by comparing the total antioxidant capacity (TAC) of shCTL and shRGS12 groups. Knockdown of RGS12 resulted in a significant decrease in TAC, indicating a decline in the cell's ability to counteract oxidative stress (Fig. 4I). To explore the effects of RGS12 knockdown on cellular function, we performed additional assays. Enzyme-linked immunosorbent assay (ELISA) revealed a significant decrease in SOD levels in the shRGS12 group compared to the control group, suggesting compromised antioxidant defense (Fig. S2A). Interestingly, GSH and NO levels remained unchanged, while MDA levels were even reduced in shRGS12 cells (Fig. S2B-D). These findings require further investigation to fully understand the interplay between RGS12 knockdown and oxidative stress markers. We next investigated the impact of RGS12 knockdown on the functional capabilities of placental trophoblast cells, including apoptosis, proliferation, hormone secretion, migration, and invasion. TdT-mediated dUTP Nick-End Labeling (TUNEL) staining revealed a significant increase in apoptosis in the shRGS12 group compared to the control (Fig. 5A and B). Similarly, CCK8 assays indicated a decrease in cell proliferation upon RGS12 knockdown (Fig. 5C). ELISA experiments showed a decrease in progesterone (P) secretion and an unchanged level of estrodiol (E2) in shRGS12 cells, leading to an increased E2-to-P ratio (Fig. 5D-F). To account for potential limitations of the HTR-8/SVneo cell line in hormone production, we verified these findings in BeWo cells, obtaining consistent results (Fig. S2E-H). Furthermore, Transwell assays and wound healing assays demonstrated a significant reduction in both migration and invasion abilities of shRGS12 cells compared to the control group (Fig. 5G-I and S2I, J). Interestingly, overexpression of RGS12 did not appear to affect proliferation, apoptosis, or mitochondrial morphology in HTR-8/SVneo cells (Fig. S3A-H).
RGS12 knockout (KO) and placenta-specific RGS12 KO mice exhibit decreased birth weight of fetuses, reduced placental efficiency, preterm birth, and placental mitochondrial dysfunction
To elucidate the in vivo role of RGS12 in pregnancy and placental function, we generated knockout mice using CRISPR-Cas9 technology. This approach targeted all exons of the RGS12 gene, resulting in global RGS12 knockout (RGS12−/−). We additionally generated a placenta-specific RGS12 knockout model by mating RGS12 floxed mice with Elf5-Cre transgenic mice. Elf5-Cre expresses Cre recombinase under the control of the Elf5 promoter, primarily active in placental trophoblast cells [39]. In this breeding scheme, Cre recombinase mediated the deletion of exons 5 and 6 of the floxed RGS12 gene, specifically within Elf5-positive cells. Immunohistochemical analysis of placentas from RGS12-edited mice at 18.5 dpc confirmed the efficiency of RGS12 knockout in both models (Fig. 6A and E). Both global and placenta-specific RGS12 knockout mice were viable and fertile. However, they exhibited reduced offspring birth weight and placental efficiency, as evidenced by a smaller pup weight to placental weight ratio, without any significant change in overall placental weight (Fig. 6B-D and F-H). These findings suggest that RGS12 deficiency may compromise placental function, impairing fetal development. Morphological examination of the gestational sac, placenta, and fetuses from placenta-specific RGS12 knockout mice at 18.5 dpc revealed earlier delivery (between 18 and 18.5 dpc) compared to controls (RGS12flox/flox). Fetuses from the RGS12flox/flox; Elf5-Cre genotype were also smaller than controls. Genotype identification was confirmed by agarose gel electrophoresis (Fig. S4A-E). Interestingly, global RGS12 knockout mice did not exhibit premature delivery, although their tolerance to low-dose LPS-induced labor was reduced, leading to earlier delivery and a significantly higher rate of preterm birth (Fig. S4G-I). This phenotypic discrepancy between the global and placenta-specific knockout models suggests a potential role for RGS12 in other tissues or the existence of compensatory mechanisms within the body. Further analysis of placentas from global RGS12 knockout mice at 18.5 dpc using hematoxylin and eosin (H&E) staining revealed a significant reduction in the labyrinth zone, while the junctional zone remained unaffected (Fig. S5A-C). Ultrasound analysis of RGS12 knockout (RGS12−/−) and wild-type (WT) mice at 18.5 dpc revealed no significant difference in fetal heart rate. However, placental umbilical blood flow velocity exhibited a dose-dependent decrease, suggesting that RGS12 deficiency may compromise placental blood flow capacity (Fig. S5D-G). This finding aligns with the observed phenotype of reduced fetal weight, potentially due to impaired nutrient and oxygen delivery to the developing fetus. Analysis of steroidogenic hormone levels in RGS12 knockout mice revealed no significant change in P levels, a significant increase in E2 levels, and no significant change in the E2/P ratio. Furthermore, the expression of key steroidogenic enzymes (CYP11A1, HSD3B1, and STS) was downregulated in these mice (Fig. S6A-F). These findings suggest that RGS12 may play a role in regulating placental steroidogenesis. In light of the in vitro data demonstrating RGS12 interaction with ATP5B and its potential role in mitochondrial function, we further investigated mitochondrial status in placentas from 18.5 dpc placenta-specific RGS12 knockout mice. As expected, p-Tyr levels were decreased in these placentas compared to controls. TEM revealed an increased number of damaged mitochondria, while the mtDNA copy number was significantly elevated. Conversely, both ATP content and total antioxidant capacity were significantly reduced (Fig. 6I-M). Collectively, these findings suggest that RGS12 deficiency disrupts placental mitochondrial function, ultimately contributing to fetal growth restriction and a potential predisposition to preterm birth in the gene-edited mice.
Restoring RGS12 expression reverses the abnormal mitochondrial-related phenotypes
To determine whether RGS12 restoration could reverse the observed mitochondrial dysfunction, we employed a series of assays in HTR-8/SVneo cells. Control cells (shCTL-CTLOE) were compared to cells with RGS12 knockdown and subsequent RGS12 overexpression (shRGS12-RGS12OE). Western blot analysis confirmed successful RGS12 restoration in the shRGS12-RGS12OE group, as evidenced by significantly increased RGS12 protein levels compared to the control group (Fig. 7A). Next, we evaluated the impact of RGS12 restoration on hormone production and oxidative stress markers. E2, P, and the E2/P ratio were measured, revealing no significant differences between the shRGS12-RGS12OE and control groups (Fig. 7A, B, and D). This suggests that RGS12 restoration does not disrupt hormonal balance. Furthermore, analysis of oxidative stress markers indicated normalized SOD activity and GSH levels in shRGS12-RGS12OE cells compared to the control group, with no significant changes in MDA or NO levels (Fig. 7E-H). These findings suggest that RGS12 restoration helps mitigate oxidative stress. To assess the effect of RGS12 restoration on mitochondrial function, we measured relative mtDNA copy number, ATP concentration, and total antioxidant capacity. The shRGS12-RGS12OE group exhibited a significant increase in all three parameters compared to the control group (Fig. 7I-K). These results highlight the crucial role of RGS12 in promoting optimal mitochondrial function. Notably, the abnormal phenotypes observed in RGS12 knockout mice also demonstrated a dose-dependent effect, indirectly supporting that restoring RGS12 expression can alleviate these abnormalities (Fig. S5D-G). Taken together, our findings substantiated that restoring RGS12 expression in HTR-8/SVneo placental trophoblast cells effectively reversed the observed mitochondrial dysfunction. This finding underscores the critical role of RGS12 in maintaining both mitochondrial function and cellular homeostasis within these cells.
PTB placentas are associated with decreased RGS12 expression and mitochondrial dysfunction
To investigate the potential link between RGS12 and PTB in humans, we collected placental tissues from patients with PTB and compared them to TB controls. Participant demographics and baseline clinical characteristics are shown in Table S2. Western blot and RT-PCR analysis revealed significantly reduced protein and mRNA levels of RGS12 in PTB placentas compared to the TB group (Fig. 8A-C). Consistent with our previous in vitro findings, PTB placentas displayed a significant decrease in mitochondrial tyrosine phosphorylation compared to controls, with no significant difference observed in cytoplasmic extracts (Fig. 8D). Furthermore, both ATP content and mtDNA copy number were significantly lower in PTB placentas (Fig. 8E and F). Analysis of steroidogenic hormone profiles in human placental tissues revealed a significant decrease in P levels in PTB compared to controls, with no significant difference in E2 levels or E2/P ratio. Interestingly, NO levels were significantly elevated in PTB placentas (Fig. 8G-J). Additionally, we observed a significant decrease in both protein and mRNA expression of ATP5B in PTB placentas, with a positive correlation between ATP5B and RGS12 expression levels (Fig. 8K-M). These findings suggest that RGS12 may regulate ATP5B expression in human placental tissues. Moreover, the downregulation of RGS12 was associated with mitochondrial dysfunction in PTB placentas, highlighting a potential correlation between these factors in human clinical samples.
Deficiency of placental RGS12 leads to increased apoptosis and activation of the p38MAPK signaling pathway
We next investigated apoptosis-related molecules in human PTB and TB placentas using Western blotting to gain further insights into the mechanisms by which RGS12 deficiency promotes trophoblast apoptosis and contributes to adverse pregnancy outcomes. We observed a significant increase in the expression of CASP9 and its activated form, cleaved-CASP9, in PTB placentas compared to controls (Fig. S7A-I). This finding was further corroborated in RGS12-knockdown HTR-8/SVneo cells (Fig. S8A-G). We next aimed to elucidate the mechanisms underlying RGS12 knockdown-induced placental dysfunction and adverse pregnancy outcomes. Transcriptomic analysis of RGS12-knockdown (shRGS12) HTR-8/SVneo trophoblast cells revealed significant changes in gene expression profiles compared to the control group (shCTL). KEGG pathway enrichment analysis identified significant enrichment of the p53 and MAPK signaling pathways (Fig. S9A-C). To validate these findings, we analyzed both clinical placental samples and shRGS12-transfected HTR-8/SVneo cells, and observed a significant enrichment of the p38MAPK signaling pathway in both groups (Fig. S10A-F). In conclusion, our findings suggest that placental RGS12 deficiency may contribute to placental dysfunction and adverse pregnancy outcomes through two potential mechanisms: 1) increased mitochondria-dependent apoptosis via the CASP9/CASP3 pathway and 2) activation of the p38MAPK signaling pathway.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}