Mice
In this study, female BALB/c background nude mice (Shanghai Experimental Animal Center, Chinese Academy of Sciences, China) from 4--6 weeks were selected to establish a PDX model. The experimental mice were housed in individual ventilated cages under specific-pathogen-free conditions. All animal care and experimental protocols were carried out according to the Chinese Animal Management Rules of the Ministry of Health. All of the procedures were authorized by the Animal Ethics Committees of Nantong University.
PDX mice were inoculated subcutaneously with patient-derived tumor tissue fragments and were used for the experiment when they passed to the F3 generation. All the mice used were age matched and were assigned randomly to groups. After the tumor volume reached 100–200 mm3, PDX mice were injected intraperitoneally (ip) with chemotherapeutic agents as indicated by the administration of cisplatin (5 mg/kg/3d), paclitaxel (5 mg/kg/3d), docetaxel (2 mg/kg/3d) or saline.
Cell culture
THP-1 and MDA-MB-468 cells were purchased from the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China). The THP-1 cells were incubated in RPMI 1640 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Gibco, Waltham, USA). The human breast cancer cell line MDA-MB-468 was maintained in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS. Both cell lines were maintained at 37 °C in an atmosphere containing 5% CO2. Both cell lines were used within 20 passages and confirmed to be mycoplasma free before use in studies.
Real-time quantitative PCR
TRIzol reagent (Invitrogen, Carlsbad, USA) was used to extract total RNA from the cell lines or tissue samples. Real-time quantitative PCR (qPCR) was performed in a total volume of 20 μL of SYBR Green PCR Master Mix (Roche, Germany). All reactions were performed in duplicate. The mRNA expression levels of the target genes were normalized to the GAPDH mRNA levels via the 2-ΔΔCT method. The primer sequences are listed in Table 1:
|
Gene
|
Primer sequences
|
|
GAPDH-F
|
5’-GCACCGTCAAGGCTGAGAAC-3’
|
|
GAPDH-R
|
5’-TGGTGAAGACGCCAGTGGA-3’
|
|
IL1-β-F
|
5’- TCCAGGATGAGGACATGAGCAC -3’
|
|
IL1-β-R
|
5’- GAACGTCACACACCAGCAGGTTA -3’
|
|
TNF-α-F
|
5’- GTTCTATGGCCCAGACCCTCAC -3’
|
|
TNF-α-R
|
5’- GGCACCACTAGTTGGTTGTCTTTG -3’
|
|
iNOS -F
|
5’- CAAGCACCTTGGAAGAGGAG -3’
|
|
iNOS -R
|
5’- AAGGCCAAACACAGCATACC -3’
|
|
CD16-F
|
5’-TTTGGACACCCAGATGTTTCAG-3’
|
|
CD16-R
|
5’-GTCTTCCTTGAGCACCTGGATC-3’
|
|
CD206-F
|
5’-CAAGGAAGGTTGGCATTTGT-3’
|
|
CD206-R
|
5’-CCTTTCAGTCCTTTGCAAGC-3’
|
|
TGF-β-F
|
5’-TGCGCTTGCAGAGATTAAAA-3’
|
|
TGF-β-R
|
5’-CGTCAAAAGACAGCCACTCA-3’
|
|
IL-10-F
|
5’-CCAAGCCTTATCGGAAATGA-3’
|
|
IL-10-R
|
5’-TTTTCACAGGGGAGAAATCG-3’
|
Western blotting
The cells or tissues were harvested in lysis buffer containing 1% phosphatase inhibitor. Total proteins separated by SDS‒PAGE were transferred to nitrocellulose membranes (Millipore, USA), which were blocked in 5% w/v nonfat milk powder in TBST (0.1 M Tris, 1.2 M NaCl, pH 7.4 and 0.1% v/v Tween 20) for 2 h at room temperature and then incubated with primary antibody overnight at 4°C. The membrane was washed with TBST adequately and incubated with the secondary antibody (Jackson, West Grove, USA). Proteins were visualized with enhanced chemiluminescence (ECL) detection reagents. The primary and secondary antibodies used were as follows: B7-2/CD86 [Unconjugated] (NOVUS, USA), CD11b (NOVUS, USA), PPARγ (Abcam, USA), IGFBP3 (Abcam, USA), monoclonal anti-β-actin (Sigma, USA), goat anti-rabbit secondary (Bioworld, USA), rabbit anti-goat secondary (Bioworld, USA), and goat anti-mouse secondary (Jackson, USA) antibodies.
Macrophage polarization
THP-1 monocytes were differentiated into macrophages by 24 h of incubation with 150 nM phorbol 12-myristate 13-acetate (PMA) (Sigma, USA) in RPMI medium. Next, the macrophages were stimulated with 20 ng/ml IFNγ (PeproTech, USA) and 100 ng/ml LPS (Sigma, USA) for 48 h to obtain the M1 phenotype. The M2 phenotype was obtained by incubation with 20 ng/ml interleukin 4 (IL-4) (Sigma, USA) and 20 ng/ml interleukin 13 (IL-13) (PeproTech, USA) for 48 h. Afterwards, the cells were collected and analyzed.
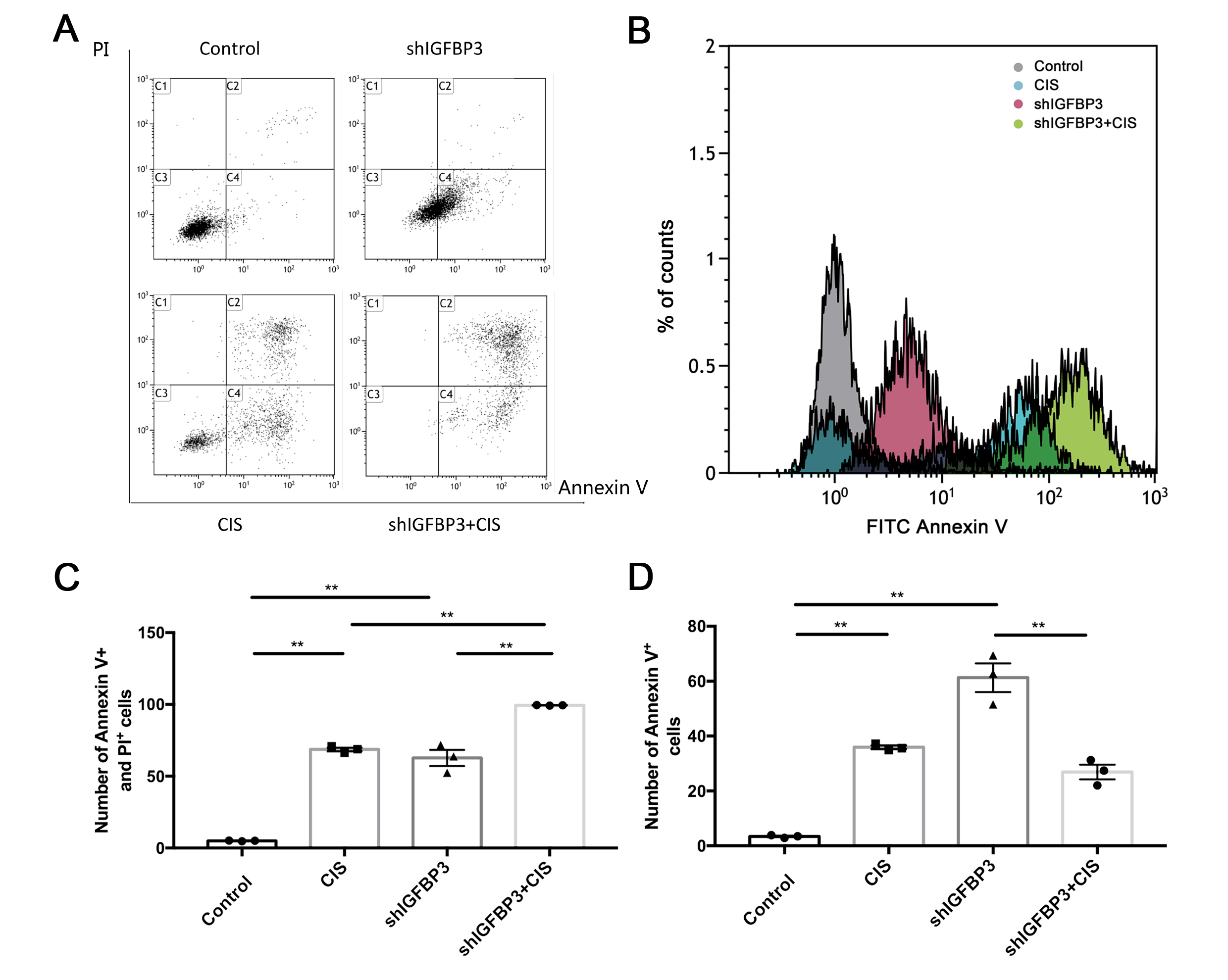
Apoptosis analysis by flow cytometry
The cells in the exponential growth phase were trypsinized and resuspended in fresh culture medium at a density of 1×105 cells/well in a 6-well plate for 24 h at 37°C. After treatment, the cells were collected with 5% trypsin, washed twice with phosphate-buffered saline (PBS), and resuspended in binding buffer at a concentration of 5×105 cells/ml. The cells (100 μl) were stained with 5 μl of fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody specific for Annexin V and 5 μl of propidium iodide (PI) (BD, USA) and then incubated in the dark at 25°C for 15 min. Subsequently, cell apoptosis was measured via flow cytometry (Beckman Coulter, USA). The data were analyzed with FlowJo 10.
Cytotoxicity assays
Cellular cytotoxicity was assessed by the level of lactate dehydrogenase (LDH) activity. THP-1 cells were seeded on inverted transwell plates, differentiated into macrophages for 24 h, washed and treated with IFNγ combined with LPS or IL-4 combined with IL-13 for 48 h. MDA-MB-468 cells were seeded on 24-well plates and incubated with polarized macrophages. After 48 h of exposure, the cocultured cells were washed for detection of LDH. LDH release was measured via a Cytotoxicity Detection Kit (Roche Diagnostics, Germany) according to the manufacturer's instructions.
Cell Counting Kit-8 (CCK-8) assay
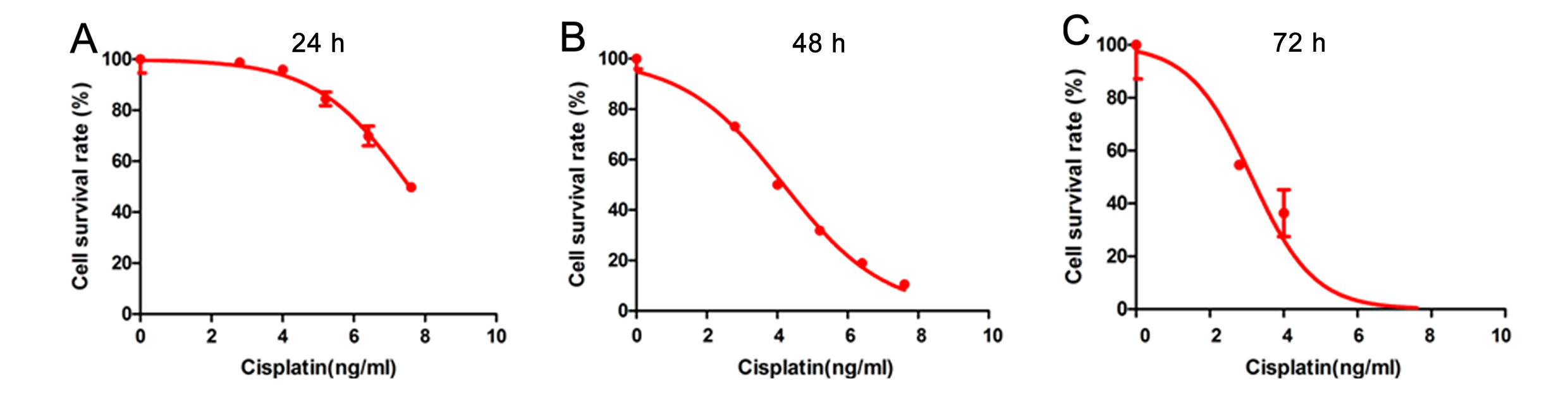
Cell viability was measured via the Cell Counting Kit-8 (CCK-8) (D Dojindo Molecular Technologies, Japan) assay following the manufacturer’s protocol. The cells were seeded into 96-well plates at a density of 3000 cells/well. After treatment with different doses of cisplatin for 24 h, 48 h or 72 h, 10 µL of CCK-8 solution was added to the cells. One hour later, the absorbance was calculated with a microplate spectrophotometer (Multiskan™ GO, Thermo Fisher Scientific) at 450 nm.
Infection with lentiviral shRNA
For the lentiviral infection experiments, recombinant lentivirus (GenePharma, China) was used according to the manufacturer's protocol. THP-1 cells were seeded at 1 × 105 cells/well in a 12-well plate and incubated overnight. The next day, control shRNA lentivirus and IGFBP3 knockdown shRNA lentivirus were combined with 10 μg/ml polybrene (Sigma, USA) and then added to the THP-1 cells. At 48 h after transduction, THP-1 cells generated with the IGFBP3 shRNA lentivirus were cultured with puromycin-containing (4 to 8 μg/ml) medium for 2 weeks to select stable clones.
Immunohistochemistry
The tumor tissue was fixed in formalin, embedded in paraffin, sectioned and heated. Subsequently, the sections were deparaffinized, rehydrated, and subjected to antigen retrieval. After being blocked with 2% goat serum, the sections were incubated with the indicated antibodies against CD11b (NOVUS, USA) and CD86 (NOVUS, USA) overnight at 4°C. The next day, the sections were washed with PBS and then incubated with the corresponding secondary antibodies for 1 hour at room temperature. The sections were treated with DAB working solution and then visualized by microscopy.
Immunofluorescence
ICC staining was performed as described previously(11). The cells were processed routinely. For ICC staining, the following antibodies were used: B7-2/CD86 [Unconjugated] (NOVUS, USA), CD11b (NOVUS, USA), PPARγ (Abcam, USA), IGFBP3 (Abcam, USA), β-Tubulin (Cell Signaling Technology, USA), Caspase 3 (Invitrogen, UK) and goat anti-rabbit secondary antibodies (West Grove, USA). Nuclei were counterstained with DAPI (Beyotime, China). Immunostaining was visualized with a confocal laser scanning microscope (SP8, Leica, Germany).
Dual-luciferase reporter assays
TheIGFBP3 promoter fragments were amplified and cloned and inserted into the pGL3 basic vector. Luciferase activity was examined via a dual luciferase assay (Promega, Madison, WI, USA) following the manufacturer's instructions.
Statistical analysis
Data analysis was conducted via GraphPad Prism 9 (GraphPad Software, San Diego, CA). The results are presented as the means ± SDs. One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used to determine significant differences between groups. For data involving only two groups, an unpaired Student’s t test was employed. P values < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01).
{kind=link}
{kind=link}
{kind=link}