All the solvents and chemicals used were purchased from Loba Cheme Pvt. Ltd., Central Drug House and Thermo Fischer Scientific, India Pvt. IR, 1H NMR and mass spectra were recorded by an Agilent Cary 630 FTIR, a Brucker Avance 400/AVIII HD 300 (FT-NMR) and Water Alliance.
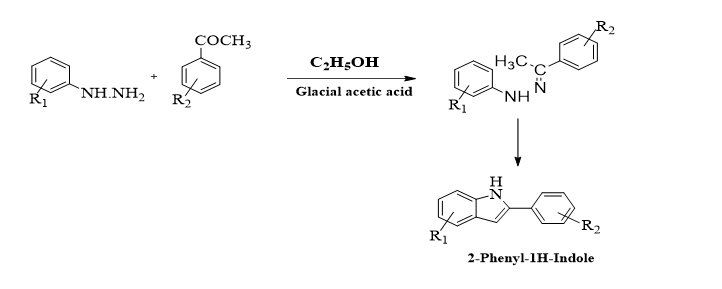
2.1 Synthesis of 2-phenylindole
2.1.1 General procedure for the synthesis of 2-phenylindole
A combination of 2.0 g (0.0167 mol) of different acetophenone derivatives and 1.8 g (0.0167 mol) of different phenyl hydrazine derivatives with 6.0 ml of ethanol and 2–3 drops of glacial acetic acid will be added and warmed. The cold reaction mixture was filtered and washed with dilute HCl, followed by the addition of approximately 12 ml of cool rectified spirit. Recrystallization was performed with ethanol to obtain pure acetophenone phenylhydrazone.
Briefly, 2.8 g of crude phenylhydrazone was added to 12.85 g (0.143 mol) of polyphosphoric acid and maintained at 100-1200C for 10–20 minutes. Cold water (32.14–40.20 ml) was added, the mixture was stirred well to obtain a complete solution of polyphosphoric acid, which was filtered and washed with water. The crude solid was refluxed with 21.42 ml of rectified spirit; a little decolorizing charcoal was added, and the mixture was filtered and washed with 40 ml of hot rectified spirit to obtain 2-phenylindole. The reaction was monitored by TLC until the completion of the reaction. Compound purity was assessed by a recoated TLC plate, and n-hexane–ethyl acetate (9:1) was used as the developing solvent. The spots were subjected to UV light, and the melting point and spectral information (FT-IR, MS, 1H and 13C-NMR) were applied to characterize all the synthesized compounds (2a to 2f).5,6, 13, 14,15,16,17 The recrystallized compound was purified by column chromatography on a silica gel 60–120 mesh (hexane/ethylacetate-90:20).
2.1.2 Synthesis of 2-(4-bromophenyl)-4chloro-1H-indole [2a]
FT-IR (cm − 1 )-NH stretching 3500 − 3100, C-H stretching 3000–3100, C = C and C-C stretching 1440–1625 and 1185–1600, N-H bending-1400-1410.
1 H NMR [DMSO-d6] δ (ppm): 6.77 (s, CH-indole, 1H), 6.81–7.55 (m, Ar-indole, 3H), 7.66–7.78 (m, Ar-CH, 4H), 11.36 (s, NH-indole, 1H).
13 C NMR [300 MHz, DMSO-d6] δ (ppm): 136.047, 132.11, 132.66, 130.06, 129.812, 129.396, 128.816, 128.563, 127.262, 114.587
MS-ESI [M+]: 306.6 (70%)
2.1.3 Synthesis of 4-chloro-2(3,4-dimethoxyphenyl)-1H-indole [2b]
1 H NMR [DMSO-d6] δ (ppm): 3.83 (d, Ar-OCH3, 6H), 6.77 (s, CH-indole, 1H), 6.81–7.55 (m, Ar-indole, 3H), 6.94–725 (m, Ar-CH, 3H), 11.36 (s, NH-indole, 1H)
13 C NMR [300 MHz, DMSO-d6] δ (ppm): 137.6, 136.912, 100.003, 125.806, 126.7, 150.3, 149.811, 109.213, 130.233, 108.398, 120.111, 111.011, 122.812, 120.812, 56.121
FT-IR (cm− 1) C-Cl (aromatic) 690, C-N stretching 1253.57, C = C (aromatic) 1464.96, C-C (aromatic) 1591.20, C-OCH3 (aromatic) stretching 2836, C-H (aromatic) 3003.75, N-H (amine) stretching 3362.96.
MS-ESI [M+]: 288.37 (86%)
2.1.4 Synthesis of 4-bromo-2(3,4-dimethoxyphenyl)-1H-indole [2c]
C-Br (aromatic) 667.97 cm− 1 C-N stretching 1348.57 cm− 1 C = C (aromatic) 1462.61 cm− 1 C-C (aromatic) 1609.20 cm− 1 C-OCH3 (aromatic) stretching 2841.57 cm− 1 C-H (aromatic) 3012.75 cm− 1 N-H (amine) stretching 3362.96 cm− 1
1 H NMR [DMSO-d6] δ (ppm): 3.83 (d, Ar-OCH3, 6H), 6.77 (s, CH-indole, 1H), 6.88 (s, CH-indole, 1H), 6.94–7.25 (m, Ar-CH, 3H), 7.50–761 (d, Ar- CH-Indole, 2H), 11.36 (s, NH- Indole, 1H)
13 C NMR [300 MHz, DMSO-d6] δ (ppm): 137.712, 137.612, 100.003, 130.406, 112.671, 150.3, 149.811, 110.113, 130.233, 108.398, 120.111, 111.011, 122.412, 120.812, 56.12
MS-ESI [M+]: 332.31 (62%)
2.1.5 Synthesis of 4-chloro-2(3-nitrophenyl)-1H-indole [2d]
C-Cl (aromatic) 771.97 cm− 1 C-N stretching 1348.57 cm− 1 C = C (aromatic) 1462.61 cm− 1 C-C (aromatic) 1609.20 cm− 1 C-NO2 (aromatic) stretching 1517.45 cm− 1 C-H (aromatic) 2918.04 cm− 1 N-H (amine) stretching 3364.96 cm− 1
1 H NMR [DMSO-d6] δ (ppm): 6.77 (s, CH-indole, 1H), 6.81–7.55 (m, CH-indole, 3H), 7.77–8.65 (m, Ar-CH, 4H), 11.36 (s, NH-indole, 1H).
13 CNMR [300 MHz, DMSO-d6] δ (ppm): 137.731, 137.611, 133.939, 131.478, 130.506, 129.474, 128.824, 124.288, 122.414, 122.164, 119.562, 119.132, 113.110.
MS-ESI [M+]: 272.32 (75%)
2.1.6 Synthesis of 4-(4-bromo-1H-indole-2-yl) benzenamine [2e]
C-Br (aromatic) 771.97 cm− 1 C-N stretching 1348.57 cm− 1 C = C (aromatic) 1462.61 cm− 1 C-C (aromatic) 1609.20 cm− 1 C-NH2 (aromatic) stretching 1517.45 cm− 1 C-H (aromatic) 2918.04 cm− 1 N-H (amine) stretching 3364.96 cm− 1
1 H NMR [DMSO-d6] δ (ppm): 6.27 (s, NH2, 2H), 6.58–7.54 (m, CH, 4H), 6.77 (s, CH-indole, 1H), 6.88–7.61 (m, Ar-CH-indole, 3H), 11.36 (s, NH-indole, 1H)
13 C NMR [300 MHz, DMSO-d6] δ (ppm): 137.612, 137.711, 100.003, 130.41, 123.900, 112.911, 145.611, 110.100, 125.600, 115.111, 128.300, 123.2, 115.111, 122.400, 128.311
MS-ESI [M+]: 289.3 (100%)
2.1.7 Synthesis of 4-bromo-2-(3-nitrophenyl)-1H-indole [2f]
C-Br (aromatic) 586.37 cm− 1 C-N stretching 1347.05 cm− 1 C = C (aromatic) 1462.61 cm− 1 C-C (aromatic) 1609.20 cm− 1 C-NO2 (aromatic) stretching 1524.03 cm− 1 C-H (aromatic) 2996.64 cm− 1 N-H (amine) stretching 3359.06 cm− 1
1 H NMR [DMSO-d6] δ (ppm): 6.77 (s, CH-indole, 1H), 6.88–7.61 (m Ar-CH-indole, 3H), 7.77–8.65 (m, Ar-CH-, 4H), 11.36 (s, NH-indole, 1H).
13 C NMR [300 MHz, DMSO-d6] δ (ppm): 137.712, 137.612, 100.003, 130.41, 123.900, 112.911, 148.421, 110.100, 136.800, 103.511, 123.233, 122.4, 129.511, 122.711
MS-ESI [M+]: 318 (72%)
2.2a Molecular docking: To determine the probable binding poses of the synthesized compound against GABA and the AMPA receptor inhibitor. The molecular docking study was carried out by Schrödinger, USA. The X-ray crystal structures were obtained from the RCSB databank (1FTL and 4COF). The ligand structure was prepared by ChemDraw professional 16.0 in .sdf. The protein was imported into a Schrödinger virtual docker, and the crystal structure was prepared by removing water and optimizing the structure by missing amino acids. The binding site of the small molecule was described by selecting the reference ligands phenytoin and phenobarbitone.17, 18
{kind=link}