Thermoplastic polyamide elastomers (TPAEs) possess remarkable characteristics such as high-temperature tolerance, superior mechanical properties, and the shape memory effect (SME). In the current study, a type of TPAEs with SME is developed by fabricating the long carbon chain polyamide (PA512) and polyethylene glycol (PEG) through a two-step melt polycondensation process. The properties of TPAEs were investigated by varying the PA512 prepolymer's molecular weight and the amount of PEG. During synthesizing TPAEs with SME, the crucial balance of COOH and OH groups was skillfully achieved by introducing biobased butanediol (BDO). The chemical structure of TPAEs is confirmed by FTIR and 1H NMR tests. By meticulously engineering the PA512 molecular weight and refining the PEG domain content, TPAEs are fabricated to elongate at a break of 592.4% at room temperature while maintaining a tensile strength of 23.1 MPa. TPAEs, which have two distinct melting temperatures, exhibit microphase separation between the PEG and PA512 domains. This phenomenon is further corroborated by the scanning electron microscope (SEM) test. Additionally, TPAEs exhibit the SME, which can fix a temporary shape when heated, twisted, and cooled, then recover to its original shape upon reheating, with TPAE230 demonstrating the most outstanding shape memory effect, achieving an average shape fixity ratio of 91.2% and a shape recovery ratio of 94.4%. This behavior is attributed to the fixing force provided by the PEG domains and the entropy elasticity of the physically cross-linked PA512 domains. The findings indicate that TPAEs exhibit enhanced SME in response to temperature changes. Leveraging this property, the development of a temperature-sensitive device holds promise for achieving breakthroughs in the realm of elastic temperature sensing applications.
Research Article
Temperature-sensitive shape memory polyamide elastomers with tunable segments: achieving excellent performances and application prospects
https://doi.org/10.21203/rs.3.rs-5191262/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 13 Nov, 2024
Read the published version in Advanced Composites and Hybrid Materials →
You are reading this latest preprint version
Thermoplastic polyamide elastomer
Shape memory polymers
Temperature-sensitive polymers
Shape memory effect
polyethylene glycol.
Bio-based polymers have recently gained increasing attention as a substitute for fossil materials due to their superior properties, including renewability and degradability, which make them more environmentally friendly and sustainable. Among them, bio-based thermoplastic polyamide elastomers (TPAEs) are a class of block copolymers that are particularly noteworthy for their exceptional chemical resistance[1], corrosion resistance[2], high-temperature tolerance[3], and outstanding mechanical characteristics[4]. TPAEs have been widely used for various applications in different fields, including automobiles, clothing, and electronic devices [5], [6], [7], [8], [9]. TPAEs are synthesized by the polycondensation of polyamide hard segments and polyether or polyester soft segments. Among these, the polyamide hard domains possess high melting and glass transition temperatures, with the physical crosslinking points in the hard domains being formed through crystallization. In contrast, the soft domains have lower melting and glass transition temperatures and are typically composed of crystalline or amorphous polymers such as polytetramethylene ether glycol (PTMEG)[10], [11], [12], [13], polypropylene glycol (PPG) [14], polyethylene glycol (PEG) [15], [16], [17], and polydioxanone (PPDO) [18]. These soft segments confer flexibility and elasticity to the polymer, while the hard segments contribute to its strength and stiffness. The thermodynamic incompatibility between the hard segments and the soft segments leads to microphase separation, which is responsible for the unique properties of the polymer. The integration of hard and soft segments in TPAEs bestows them with an array of exceptional properties, including customizable chemical structure, shape memory effect (SME)[19], remarkable electrical characteristics[20], and self-healing abilities[21]. Shape memory polymer (SMP) typically comprises two phases: one is responsible for maintaining the original shape, while the other provides the fixing force to retain the temporary form. The microphase separation structure within TPAEs enables the SME to occur. Shibasaki[16] has successfully developed a range of shape memory polymers (SMP) through the conventional solution polycondensation process, utilizing N-methylaminobenzoic acid (MAB)-based oligomers and PEG. The remarkable properties of the PEBA films, with a shape fixity ratio of 91% and a shape recovery ratio of 97%, effectively demonstrate the potential of their temperature memory functionality.
The general approach for synthesizing TPAEs involves first preparing end-carboxylated polyamide prepolymers, followed by their reaction with an equimolar amount of polyester or polyether [22], [23], [24], [25]. During the adjustment of soft and hard segment compositions, a frequent occurrence of an imbalance between COOH and OH groups often leads to a decrease in the reaction rate. To address this issue, the Peyravi group[26] developed a method for fabricating TPAEs using carboxy-terminated PA6 and polyethylene oxide (PEO) as the primary constituents. They incorporated adipic acid (AA) and hexamethylenediamine (HMDA) to achieve an accurate equilibrium of COOH and OH groups. AA serves as a chain extender to react with PEO, during which the excess OH is consumed when the PA6 hard segment content is as low as 10 wt%. Correspondingly, HMDA functions as a chain extender to react with carboxy-terminated PA6, where the excess COOH is utilized when the PA6 hard segment content is as high as 80 wt%. The crux of this strategy lies in employing the chain extension agent to neutralize the surplus carboxy or hydroxy groups, thus ensuring a stringent stoichiometric balance of COOH and OH components. However, HMDA is not conducive to the industrial production of TPAEs due to its prohibitive cost and inherent toxicity. Yuan[27] decreased the quantity of PTMG by incorporating a small amount of ethylene glycol (EG) to offset the absence of the OH component. This adjustment led to an accurate balance of COOH and OH, achieving an accurate equilibrium. In summary, the research by the Yuan group and Peyravi group offers an approach for achieving a stoichiometric balance of terminal functional groups by incorporating substances with specific end groups. Nevertheless, for industrial melt polymerization processes, EG has a boiling point of 197.3°C—significantly lower than the melting temperature of PA512 at 203.1 ℃. As such, it vaporizes before reaching the melt polymerization temperature, presenting challenges in the development of TPAEs with high melting points. Furthermore, the raw materials for polyamide production originate from petroleum, which is increasingly subject to concerns over depleting petroleum resources and energy shortages. As such, petroleum-based TPAEs are falling out of favor[28], [29]. In recent years, bio-based materials have emerged as a more sustainable alternative, making the production of bio-based TPAEs a pressing matter for domestic manufacturers[30], [31]. However, butanediol (BDO) is a widely used novel bio-based material for the synthesis of copolyesters[32] which has a higher melting temperature of 228.2 ℃. For TPAEs, which also undergo esterification reactions, it is proposed to substitute EG with BDO to produce TPAEs with varying ratios of soft segments through melt polymerization at high temperatures[33], [34].
A series of TPAEs with excellent tensile strength and elongation at break were successfully prepared by controlling the content of PEG and using BDO to offset the absence of the OH component. The structures of the TPAEs were systematically characterized using nuclear magnetic resonance (NMR), Fourier transform infrared spectroscopy (FTIR), wide-angle X-ray diffraction (WAXD), and differential scanning calorimetry (DSC). Furthermore, the shape memory effect of the TPAEs was investigated, and TPAE230 exhibited an impressive shape fixation rate of 91.2% and a remarkable shape recovery rate of 94.4%. Compared with other thermoplastic SMPs, PEG-based TPAE has a water vapor barrier and CO2 barrier properties. [35]. The newly developed polymers possess excellent dimensional stability at elevated temperatures, significant tensile properties, and good SMEs, making them highly promising for future applications in sports shoes, wearable devices, and soft actuators[36], [37].
2.1. Materials
Tetrabutyl titanate (Ti (OC4H9)4) and Trifluoroacetic acid-d were purchased from Aladdin Bio-Chem Technology Co. Ltd (Shanghai, China). Pentanediamine (PDA) and dodecanedioic acid (DDA) were purchased from Cathay Biotech Co. Ltd (China). Polyethylene glycol (PEG, Mn = 2000 g mol− 1) was purchased from Jiangsu Haian Petrochemical Co. Ltd (Jiangsu, China). All the chemicals were used as received without any further treatments.
2.2. Synthesis of Bio-based thermoplastic polyamide elastomer based on PA512 and PEG
2.2.1 Step 1: Synthesis of Bio-based PA512 with binary carboxyl end-groups (Pre PA512)
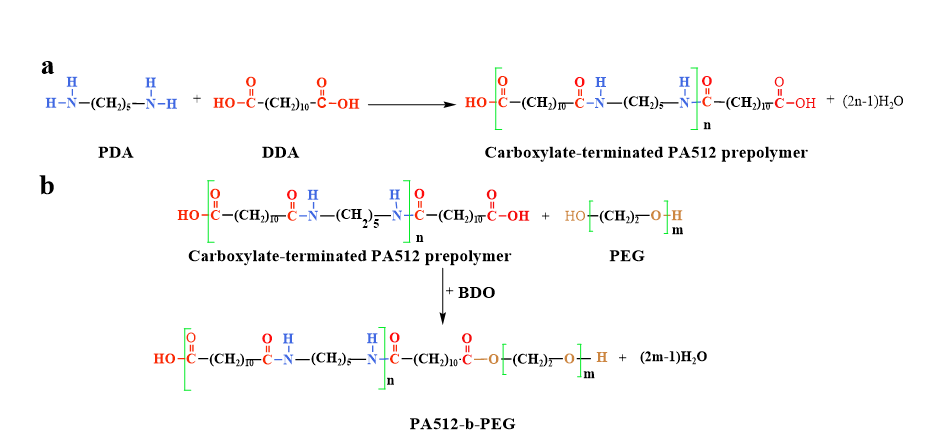
The carboxyl-terminated PA512 was synthesized through polycondensation of PDA and DDA at a calculated mass ratio (Scheme 1, a). The feeding mass ratio for the PA512 prepolymer of different molecular weights is listed in Table 1. The equal molar masses of DDA and PDA were first mixed well and then transferred into a 5 L stainless-steel batch tank reactor, followed by the addition of some distilled water and DDA as a capping agent. The gas in the reactor was replaced with nitrogen and kept at 0.2 MPa pressure to ensure that the polycondensation took place in an inert gas environment. Subsequently, the prepolymer was synthesized according to the following procedure: I). The stirrer was turned on when the mixture was raised to 80°C, and the pressure in the reactor was maintained at 0.2 MPa for 0.5 h. II). The vent valve was opened when the mixture was heated to 180°C at 1.5 MPa. III). The temperature was increased to 240°C, and the pressure was maintained at 2.0 MPa in the reactor for 1 h. IV). Release the pressure of the reactor to atmospheric pressure; the mixture was allowed to react for an additional 1 h under these conditions. IIV). The mixture was allowed to react for an additional 0.5 h under vacuum conditions. When the reaction was complete, the prepolymer was dried and successfully synthesized with different molar weights (2000, 3000, and 4000 g mol− 1) in the present work. The experimental average molecular weight of the PA512 prepolymer was determined by the viscosity test, and it is in good agreement with Eq. (1).
$$\:<{\text{X}}_{\text{n}}\ge\:(1+r)/(1-r)$$
1
where < Xn> represents the number of structural units of the PA512, and r represents the mole ratio of PDA to DDA.
2.2.2 Step2: Synthesis of Thermoplastic PA512-b-PEG
The TPAE was synthesized through a well-balanced process that involved the esterification of the PA512 prepolymer and PEG (Scheme 1, b). The addition of BDO served to balance the deficient OH for alcohol-acid equilibrium. To synthesize TPAEs, the following methodical procedure was adopted: PA512 prepolymer (2000, 3000, and 4000 g mol− 1), PEG (2000 g mol− 1), BDO, and Ti (OC4H9)4 were introduced into a 5 L high-pressure reactor. The reactor was then purged of air and filled with N2 at 0.25 MPa, and the temperature was raised to 220°C for a duration of 2 h. II) The N2 was evacuated, and the high-pressure reactor was heated to 260°C. The mixture was allowed to undergo further reactions for 2 h under vacuum conditions. III) Once the reaction was complete, the prepolymer was dried, and a series of TPAE block copolymers with varying compositions were successfully synthesized. These copolymers were designated as TPAExy, where x and y represented the molar weight of PA512 prepolymer (2000, 3000, and 4000 g mol− 1) and the mass fraction of PEG. In this work, a diverse array of TPAE block copolymers, including TPAE410, TPAE420, TPAE430, TPAE440, TPAE330, and TPAE230, were synthesized for comprehensive characterization and investigation. As an example, TPAE430 was synthesized by the PA512 prepolymer at 4000 g mol− 1 and PEG at 30 wt% of the total polymer mass.
2.3. Characterizations
The intrinsic viscosity [η] of the samples was measured using a Ubbelohde viscometer in a thermostated water bath maintained at 25 ± 0.1°C in a 96 wt% sulfuric acid solution of TPAEs with a concentration of 0.01g mL− 1. The intrinsic viscosity [η] was calculated using the Solomon-Ciuta equation of the single-point method (Eqs. (2) and (3)).
$$\:{\eta\:}_{sp}={\eta\:}_{r}-1=(\text{t}-{\text{t}}_{0})/{\text{t}}_{0}$$
2
$$\:\left[\eta\:\right]=\sqrt{{2(\eta\:}_{sp}-\text{ln}{\eta\:}_{r})}/\text{c}$$
3
where ηsp represents the specific viscosity, ηr represents the relative viscosity, and c represents the solution concentration (g mL− 1), respectively.
The FTIR spectra of TPAEs were recorded on a Tensor-27 instrument (Bruker, Ettlingen-Erman) spectrometer at room temperature. The test mode was attenuated total reflection (ATR), and the scanning range was 4000–500 cm− 1. 1H NMR spectra of TPAE block copolymers were also obtained at room temperature on a Bruker DPX-400 spectrometer, and trifluoroacetic acid-D was used as the solvent (δ = 11.50 ppm).
The thermal properties of the TPAEs were evaluated using DSC and thermogravimetric analysis (TGA). 5 ± 0.3 mg of sample was used to conduct the DSC analysis on a Mettler-Toledo instrument at a heating and cooling rate of 10°C/min under a nitrogen atmosphere. The first cooling crystallization curve and secondary heating melting curve were obtained in the temperature range of -50 to 250°C after the samples were heated up to 250°C and kept at this temperature for 5 minutes to eliminate the thermal history. 5–8 mg of sample were used to carry out the TGA on a Mettler-Toledo TGA instrument. The TGA analysis was performed in the nitrogen atmosphere with a heating rate of 10°C/min from 50 to 600°C. XRD spectra of TPAE block copolymers (2 mm thickness) were recorded on a Rigaku XRD Ultima IV X-ray diffractometer using Cu Kα (0.154 nm) as a radiation source with a scanning range of 2θ = 5–50 ° and scanning speed of 3 ° min− 1.
The microscopic structure of different TPAEs was observed using a field-emission scanning electron microscope (SEM, JSM-7200F, Japan). Adoption of low-temperature fracture for TPAEs, where the sample is first frozen in liquid nitrogen for four hours and then interrupted with the pendulum of impact testing. Before the SEM measurement, all specimens were sputter-coated with gold. Dynamic mechanical analysis (DMA) was performed in the small stretching mode with a Q800 instrument at a frequency of 1 Hz with sample dimensions of 25 mm (length) × 4 mm (width) × 2 mm (thickness). The samples were heated from − 70 to 150°C at a heating rate of 3°C/min. The storage modulus (G'), loss modulus (G"), and loss factor (tan 𝛿) were determined as a function of temperature from the curves.
The mechanical properties of the TPAEs were investigated at room temperature by performing tensile tests on a universal testing machine (SANS-CMT6104). The dumbbell-shaped specimens (4 mm width and 2 mm thickness) with an initial fixture distance of 25 mm were stretched until the break at a cross-head rate of 10 mm min− 1. The stress-strain curves were obtained, and the mechanical properties, including yield stress, ultimate stress, and elongation at break, were determined from the curves.
The rheological properties of the TPAEs were evaluated using an ARES-G2 rotational rheometer. The instrument was operated with a sample thickness of 2 mm, a measurement temperature of 210°C, and a frequency range extending from 0.01 to 100 Hz. The energy storage modulus (G'), loss modulus (G''), loss factor (tan δ), and complex viscosity (η*) were determined as a function of angular frequency for various samples.
2.4. Shape Memory Effect
To quantitatively characterize the SME, bending-recovery experiments were conducted on the sample. The heat-triggered shape memory behavior of TPAE was investigated using samples with dimensions of 75 mm (length) × 5 mm (width) × 2 mm (thickness). The samples were initially fixed into a "U" shape at 60°C and then rapidly cooled down to -30°C to set the "U" shape. The external force was removed, and the resulting angle was recorded (θ0). Subsequently, the temperature was raised back to 60°C, whereupon the samples exhibited shape recovery. The entire recovery process was captured using a digital video camera. The shape fixity ratio (Rf) and shape recovery ratio (Rr) are two important metrics used to characterize the SME. They are given by:

Where θ0 represents the initial deformation angle at a time equal to 0 seconds, θt signifies the angle at a subsequent time point of t s. The whole process was recorded, and Rr and Rf could be calculated.
Meanwhile, the macroscopic shape memory effect was observed through a series of precise steps. The TPAE, measuring 75 mm in length, 5 mm in width, and 2 mm in thickness, was subjected to vacuum drying at 80°C for a duration of 4h. Subsequently, the sample was twisted into a helical shape after being heated to 60°C. This helical configuration was maintained by cooling the sample down to − 30°C and maintaining this temperature for 5 min. Recovery of the original shape was initiated upon heating the sample to 60°C again, with the reshaping process recorded using a camera.
3.1. Chemical Structure and Intrinsic Viscosity of TPAEs.
The viscosity-average molecular weight of the TPAE and PA512 was calculated through Eq. (3), and the results are presented in Table 1. An increase in characteristic viscosity serves as a critical indicator of the successful synthesis of TPAE. The structure of the TPAEs was initially characterized using FTIR (Fig. 1). The peak observed at 1644 cm− 1 was ascribed to the C = O stretching vibration of the amide group, while the peaks at 3320 and 1530 cm− 1 were identified as the N-H stretching and bending vibrations of the amide group. The appearance of these characteristic peaks confirmed the successful synthesis of PA512.
Additionally, new characteristic peaks appeared at 1735 and 1106 cm− 1 for all TPAEs. The 1735 cm− 1 peak was attributed to the symmetric telescopic vibration of C = O in the ester moiety, while the 1106 cm− 1 peak corresponded to the anti-symmetric telescopic vibration of C-O-C in the PEG segment. These observations indicated that the esterification reaction between the PA512 prepolymer and PEG had occurred, successfully synthesizing the TPAE block copolymer.
|
sample |
PDA/g |
DDA/g |
PEG/g |
BDO/g |
PA512[η]a |
TPAE[η]b |
|---|---|---|---|---|---|---|
|
TPAE410 |
187.22 |
456.66 |
66.67 |
14.74 |
25.62 |
37.51 |
|
TPAE420 |
187.22 |
456.66 |
150.00 |
9.45 |
25.62 |
36.51 |
|
TPAE430 |
187.22 |
456.66 |
257.14 |
2.70 |
25.62 |
43.06 |
|
TPAE440 |
187.22 |
456.66 |
400.00 |
0 |
25.62 |
28.61 |
|
TPAE330 |
187.22 |
469.70 |
266.34 |
9.32 |
20.36 |
38.27 |
|
TPAE230 |
187.22 |
499.07 |
287.03 |
17.22 |
15.32 |
37.46 |
a The intrinsic viscosity of PA512prepolymer.
b The intrinsic viscosity of TPAEs.
In addition, as shown in Fig. 2, the signals at δ = 1.70 (H3) and δ = 2.11 (H2) are attributed to the protons connected to the amide bond of the PA512 segments, respectively. The signals at δ = 3.19 (H1) and δ = 4.30 (H11) represent the protons connected to the − NH − and C = O of the PA512 segments, respectively. The signal at δ = 4.30 (H11) represents the protons connected to the ester bond of the PEG segments. The results of FTIR and 1H NMR analysis provide evidence for the presence of amide and ester groups, which indicates that the TPAEs were successfully synthesized.
3.3. Thermal Properties and Crystallization Behavior
TGA was conducted to evaluate the thermal stability of TPAEs and PA512 under a nitrogen atmosphere, and the corresponding curves are presented in Fig. 3 and Table 2. Figure 3 presents the TG and DTG curves for TPAEs and PA512, while Table 2 provides the corresponding thermal decomposition data. As depicted in Fig. 3. a and 3. b, for TPAEs, the values for the thermogravimetric parameters T5%, T50%, and Tmax decreased as the proportion of PEG increased in the elastomer from 413.4°C, 462.5°C, and 470.2°C to 375.1°C, 431.3°C, and 430.6°C, respectively. This observation is attributed to the increased presence of PEG groups, which disrupted the regularity of the molecular chain within the nylon crystal region[15]. This, in turn, decreases the crystal density per unit volume of TPAE and reduces the intramolecular hydrogen bonding strength, leading to decreased crystallinity and, consequently, a decreased thermal stability performance for TPAE. Figure 3.c and Fig. 3.d exhibit that when the PEG content in the elastomer is fixed, the T50% and Tmax values of TPAE show an increasing trend with the rise of the molecular weight of the hard segment, ranging from 434.2°C to 442.3°C and 430.6°C to 463.5°C. This observation can be primarily attributed to the fact that as the molecular weight of the hard segment rises, the crystalline region of the nylon phase becomes more stable, and the impact of PEG becomes relatively insignificant compared to the nylon phase. Thus, the crystal density per unit volume of TPAE rises while the crystallinity decreases, leading to a decrease in the thermal stability of TPAE. However, when the crystal density per unit volume increases, the density of intramolecular hydrogen bonding also increases, leading to an increase in the crystallinity and, consequently, an improvement in the thermal stability of TPAEs.
|
Sample |
T5%(℃) |
T50%(℃) |
Tmax(℃) |
Remant(%) |
|---|---|---|---|---|
|
PA512 |
413.4 |
462.5 |
470.2 |
0.6 |
|
TPAE410 |
392.4 |
450.9 |
468.5 |
0.8 |
|
TPAE420 |
390.5 |
447.4 |
466.4 |
1.1 |
|
TPAE430 |
386.7 |
442.3 |
463.5 |
1.2 |
|
TPAE440 |
375.1 |
431.3 |
430.6 |
1.6 |
|
TPAE330 |
384.8 |
440.6 |
439.3 |
1.8 |
|
TPAE230 |
384.3 |
434.2 |
430.5 |
1.2 |
DSC curves of different TPAEs and PA512 during the first cooling and second heating scans are shown in Fig. 4, and the corresponding thermal properties are summarized in Table 3. The TPAE block copolymer is composed of PEG soft segments with lower polarity and PA512 hard segments with high polarity. The polarity mismatch between the hard and soft segments gives rise to microphase separation in the TPAEs block copolymer, resulting in two distinct melting and crystallization temperatures[16]. As illustrated in Fig. 4.a, as the PEG content increased from 0–20%, the melting and crystallization temperatures of TPAEs decreased from 203.1°C to 196.5°C and from 152.2°C to 147.8°C, respectively. This phenomenon can be attributed to the disruption of the crystalline zone structure of the PA512 phase upon the introduction of PEG, which disturbs the molecular chain arrangement within the crystalline zone of the PA512 phase, leading to a reduction in the crystallinity of the PA phase. Concurrently, the density of hydrogen bonding in the elastomer decreases, causing a weakening of the intermolecular forces and making the chain forging more susceptible to movement. This results in a further decrease in the crystalline density of the crystal zone, a reduction in crystallinity, and an increase in the degree of disorder in the elastomer's structure. The emergence of a micro-phase separation structure and a decrease in the melting temperature are also observed. When the PEG content was increased to 30%, the TPAE block copolymer still exhibited two distinct crystallization peaks and melting peaks. However, at this point, the crystallization and melting temperatures of the PA512 segment were similar to those of the PA512 prepolymer. It was presumed that this occurred due to the agglomeration of a large amount of PEG, which increased the degree of micro-phase separation[38].
Therefore, we also synthesized TPAE230 and TPAE330 for comparison with TPAE430. As shown in Figs. 4. c and 4. d, when the content of the soft segment is constant, the melting point of the PA segment in the elastomer decreases from 204.5°C to 200.1°C with a decrease in the molecular weight of the hard segment. This can be attributed to the reduction in the density of intermolecular hydrogen bonding and the weakening of the intermolecular force when the molecular weight of the hard segment decreases. As a result, the "pulling" effect of the soft segment on the hard segment becomes more pronounced, causing the PA512 phase to exist as a discrete phase within the PEG matrix, giving rise to a micro-phase separation state. This leads to a gradual decrease in the melting temperature[39].
|
Sample |
Tc,s(℃) |
Tm,s(℃) |
Tc,h(℃) |
Tm,h(℃) |
ΔHc,h(J/g) |
ΔHm,h(J/g) |
ΔHc,s(J/g) |
ΔHm,s(J/g) |
|---|---|---|---|---|---|---|---|---|
|
PA512 |
— |
— |
152.2 |
203.1 |
59.1 |
62.3 |
— |
— |
|
TPAE410 |
12.7 |
37.2 |
150.5 |
198.5 |
45.5 |
58.9 |
7.4 |
4.6 |
|
TPAE420 |
16.2 |
38.5 |
147.8 |
196.5 |
38.5 |
56.1 |
9.8 |
9.5 |
|
TPAE430 |
39.2 |
53.5 |
155.7 |
204.9 |
36.4 |
35.6 |
36.0 |
34.1 |
|
TPAE440 |
32.8 |
50.5 |
154.3 |
204.5 |
33.3 |
35.6 |
52.8 |
53.3 |
|
TPAE330 |
23.17 |
40.7 |
168.2 |
205.5 |
27.1 |
28.2 |
15.4 |
15.1 |
|
TPAE230 |
25.33 |
41.8 |
165.3 |
200.1 |
38.2 |
45.1 |
24.7 |
30.4 |
Since block copolymers usually have two or more components, the molar weight and crystallizability of different components can affect the crystallinity of different domains in the block copolymer[40]. WAXD analysis at 25 ℃ was performed to study the crystal structure of TPAEs (Fig. 5). The XRD pattern of PA512 exhibits a single diffraction peak at 2θ = 20.9 °, which is attributed to the α-crystalline structure. When the molecular weight of the hard segment is held constant, the addition of soft segment mass results in the appearance of new diffraction peaks at 2θ = 19.1 ° and 2θ = 23.2 ° for TPAE, which are attributed to the α-crystalline structure. TPAE410 and TPAE420 do not exhibit obvious crystallization peaks at these two positions at this time. This is because the crystallization temperatures of the soft segments are 12.7°C and 16.2°C, respectively, so the soft segments of TPAE410 and TPAE420 are only partially crystallized or amorphous at 25°C, which explains why no obvious diffraction peaks were observed on the XRD pattern. For TPAE430 and TPAE420, both the γ and α crystalline forms are present. This is because the addition of PEG leads to a more flexible elastomer with a higher molecular weight, allowing the chain segments to move more freely and facilitating crystallization, resulting in the formation of both γ and α crystals.
3.3. Phase Morphology
SEM analysis was performed to ascertain the surface morphologies of the TPAEs and PA512, by which the phase structure of the polymer, especially the microphase separation structure, can be observed. In the present work, we can observe the distribution of the microphase domains of TPAE330 and TPAE430 using an SEM microscope (Fig. 6). It can be observed from Fig. 6.a that PA512 possesses a continuous phase with a relatively flat and smooth overall morphology. However, the introduction of PEG leads to the elastomer's interface becoming rough. When the polarity difference between the hard and soft segments is significant enough, the elastomer exhibits a micro-phase separation structure, as seen in TPAE420, TPAE430, and TPAE330, which show the micro-phase separation. TPAE330 exhibits the most prominent micro-separation, contributing to its largest elongation at break.
The DMA analysis serves as additional validation of the microphase separation structure in the polymer, corroborating the trends observed in DSC. Examining the tan δ-temperature curves (Fig. 7. b and Fig. 7. d), it is evident that TPAE exhibits two transition peaks, which correspond to the α and β transitions, respectively, transitioning from higher to lower temperatures[41]. The α transition signifies the commencement of movement within the PA512 hard segments in the amorphous region, triggered by the dissociation of hydrogen bonds among PA512 molecular chains. The temperature of this α transition corresponds to the Tg of the PA512 amorphous phase, which can serve as an additional switching temperature to enable a multi-shape memory effect. On the other hand, the β transition is associated with the synergistic effect of two distinct relaxations: one originating from the relaxation of the amorphous PEG soft segments, and the other attributed to the local chain mobility of amide groups within the amorphous PA512 hard domains that do not participate in hydrogen bonding with each other. These findings are in agreement with the observations made in Fig. 7.
Indeed, Fig. 7. d presents a clear visualization of this trend: when the molecular weight of the soft segment remains constant, the α transition temperature gradually shifts towards lower temperatures and converges towards the β peak as the molecular weight of the PA512 hard segment decreases. This observation suggests improved compatibility between the PA512 hard segments and the PEG soft segments, thus confirming the presence of microphase separation. When the content of PEG was increased to 20 wt%, the α transition temperature shifted towards lower temperatures and moved closer to the β peak. This suggested that as the molecular weight of PEG increased, the molecular polarity difference between PA512 and PEG segments became greater, resulting in inhomogeneous dispersion of the bi-continuous structure, leading to a reduction in microphase separation. However, when the PEG content was increased to 30 wt%, the α transition temperature inclined towards the temperature of nylon, and the degree of microphase separation increased, as can be seen in the SEM image. This indicated that a higher content of PEG could lead to more significant microphase separation.
Figure 7. a and Fig. 7. c display the storage modulus as a function of temperature. Below the Tg, the storage modulus remains relatively constant with increasing temperature due to the freezing of chain segments. As depicted in Fig. 7. a, the E′ value decreases continuously with an increase in PEG content. Particularly around − 30°C, the E′ begins to decrease sharply, attributable to the glass transition of PEG. Above Tg, the storage modulus decreases dramatically due to the mobilization of chain segments. At approximately 55°C, a relatively stable rubbery plateau region becomes evident in all samples, indicating a highly elastic state that is characteristic of thermoplastic elastomers and block copolymers with uniform PA512 segments.
3.4. Tensile properties analysis
The tensile properties of TPAE block copolymers were measured at room temperature using a SANS-CMT6104 tensile testing machine. All samples exhibit a typical stress-strain curve of a thermoplastic elastomer, further confirming that the introduction of PEG transforms the PA512 into an elastomer. All the samples present a yield behavior during the stretching process, which means polymer chain slipping occurs, resulting in plastic deformation when the external force exceeds the elastic limitation of the material.
As shown in Fig. 8. a, when the molecular weights of PEG and PA512 are kept constant, the yield strength (σy) is 42.3 MPa, 29.2 MPa, 24.0 MPa, and 31.0 MPa, respectively, while the elongation at break (εb) increases as the proportion of PEG increases. This can be attributed to the fact that as the content of PEG segments in the elastomers grows, the PEG segments reduce crystallinity and the force between the molecular chains of the hard segments, resulting in a decreased rigidity of the elastomers and allowing for greater elongation. Therefore, the tensile strength decreased. Concurrently, the elongation at the break of the elastomer increased significantly, showcasing favorable toughness, which is attributable to the integration of polyethylene glycol. The latter enhances the randomness of the molecular chain, decreases intermolecular polarity, and notably enhances the flexibility of the molecular chain. However, the TPAE440's injection-molded samples displayed a skin-core separation structure. Under tensile force, the samples were pulled apart directly at the narrow neck following the yield point, potentially due to the high PEG content causing part of the PEG to agglomerate. The tensile strength of TPAE430 is indeed higher than that of TPAE420, which seemingly contradicts the general understanding that the tensile strength of elastomers decreases when the content of soft segments increases. One possible explanation for this phenomenon could be the presence of butanediol in TPAE420, which introduces more low molecular chain segments and disrupts structural regularity. Conversely, TPAE430 has a greater density of hydrogen bonding, resulting in stronger intermolecular forces and, therefore, a macroscopically superior tensile strength[42]. As for TPAE440, the decrease in its tensile strength can be attributed to the use of PEG as the soft segment without incorporating BDO groups. Thus, the molecular chain is more rigid and regular, with increased chain flexibility leading to decreased macroscopic tensile strength. Furthermore, the integration of PEG into the PA512 phase renders it more susceptible to stress concentration phenomena, leading to the development and enlargement of microcracks. As stretching persists, these microcracks continue to expand until the sample fails macroscopically, leading to a decrease in elongation at break.
Figure 8. b shows the yield strength (σy) increases and elongation at break (εb) decreases with the molecular weight of PA512 increasing when the molecular weight of PEG remains the same. This phenomenon occurs because as the molecular weight of the hard segments decreases, the hydrogen bonding density of TPAE reduces, leading to weaker intermolecular forces and a lower σy.
3.5. Rheological properties
The unique dynamic fluidity of the polymer melt is due to its chain-like development structure, and understanding this fluidity is crucial for analyzing the polymer structure and compatibility[43]. As shown in Figs. 9. a and 9. b, the storage modulus (G') of TPAE increases significantly with increasing frequency. This is because, in the low-frequency region, the deformation of the molecular chain can keep up with the changes in the external force, resulting in a lower storage modulus (G'). However, as the frequency increases, the acting time of the shear force becomes shorter, which means the deformation time of the macromolecular chain also becomes shorter, far less than the relaxation time of the molecular chain. This leads to the deformation of the macromolecular chains not being able to keep up with the changes in external forces, resulting in the system's elastic properties becoming dominant, causing G' to increase significantly.
As shown in Figs. 9. a and 9. d, the storage modulus (G') gradually increases with the increase in PEG content. The addition of PEG enhances the interaction between TPAE, hindering the movement of molecular chains and making the conformational change more complex. Under the action of external forces, more energy can be absorbed, leading to an increase in the storage modulus (G'). Figure 4(c) presents the curve of complex viscosity (η*) of TPAE as a function of angular frequency at 210.0°C. It can be observed that the complex viscosity of the polymer decreases with the increase in angular frequency, which is a typical shear thinning phenomenon. Under the action of high shear frequency, the disentanglement is greater than the entanglement, leading to the destruction of the internal network structure of the system, a decrease in viscosity, and the occurrence of shear thinning. Additionally, as the content of PEG increases, the complex viscosity of the elastomer gradually trends towards that of pure nylon. This can be attributed to the introduction of PEG, which leads to the originally stable system becoming more chaotic with increased molecular chain entanglement, resulting in increased polymer viscosity. Therefore, the complex viscosity gradually increases and ultimately converges to a point.
As shown in Figs. 9. d and 9. f, when the molecular weight of the hard section increases, both G' and G'' exhibit an increasing trend at low frequencies, while converging to one value at high frequencies. The storage modulus of the polymer is influenced by the molecular weight distribution at high frequencies while the change in storage modulus at low frequencies is dictated by the molecular structure. As the molecular weight of the hard segments grows, the hydrogen bonding density within the elastomer increases, resulting in enhanced intermolecular forces. This leads to an increase in the energy G' and a decrease in η* at low frequencies.
3.6. Shape memory performance and Temperature sensing device
There are several key elements that SMPs must possess to exhibit the shape memory effect [44], [45], [46], [47]: First, the soft and hard segments must undergo adequate phase separation to achieve an optimal SME. Secondly, cross-linking points must be formed through chemical or physical interactions between the hard segments, allowing the polymer to maintain its original macroscopic shape while preventing chain slipping and breaking and providing resilience through an entropy-increasing process during deformation. Lastly, the crystallization of soft segments immobilizes the polymer chains, fixing the temporary shape when the temperature falls below the soft domains' crystallization temperature. In the present study, the highly polar PA512 hard segments and nonpolar PEG soft segments underwent microphase separation due to thermodynamic incompatibility. Moreover, the PA512 hard segments formed crystals acting as physical cross-linking points through hydrogen bonding, driving the material to recover its shape above the Tm of PEG. The PEG soft segments, on the other hand, provided a fixed force for deformation through crystallization below the Tc of PEG. Therefore, the TPAE block copolymer demonstrated an outstanding SME.
The heat-triggered shape memory behavior of TPAE block copolymers was investigated, and the results are presented in Figs. 10. a-c. Figure 10.a illustrates the complete shape recovery process of TPAE420 at 60°C, where the sample transitions from the temporary "U" shape to its permanent "bar" shape within 210 s. Figure 10.b depicts the impact of PEG content on the recovery ratio of the composites. It was observed that as the amount of PEG increased, the recovery ratio of the composites also improved, reaching 69.4% and 73.3% for PEG contents of 20 wt% and 30 wt%, respectively. This can be attributed to the increase in the proportion of PEG soft segments acting as reversible microdomains within the TPAE matrix, which provides a larger restraining force for deformation, ultimately leading to enhanced shape memory effects in the TPAE system. Figure 10.c reflects the effect of the PA512 prepolymer molecular weight on the recovery ratio of the TPAE. The increasing molecular weight of PA512 prepolymer leads to an increase in cross-linking points, which results in a gradual increase in the recovery ratio of up to 94.4%, 90.1%, and 73.3%. However, this also causes the TPAE restitution rate to decrease as the higher molecular weight produces more cross-linking points within the TPAE block copolymers. Additionally, irreversible plastic deformation occurs more readily, contributing to the improved shape-memory effect. The insufficient fixing force during the shape programming step, which is attributed to the crystallization of PEG domains, is unable to counteract the elasticity of the PA512 crystalline network, failing shape fixation. The Rf and Rr were recorded in Table 4.
|
Sample |
Rf (%) |
Rr (%) |
|---|---|---|
|
TPAE420 |
79.5 |
69.4 |
|
TPAE430 |
62.4 |
73.3 |
|
TPAE330 |
90.7 |
90.1 |
|
TPAE230 |
91.2 |
94.4 |
The TPAE420 and TPAE330 samples demonstrated macroscopic shape memory behavior (Fig. 11). Both samples were twisted after being heated to 60 °C and then placed in a -30 °C refrigerator for 5 min to fix the helical shape. Upon reheating to 60 °C, the sample almost completely recovered to their original shapes, showcasing their excellent shape memory performance. It is worth highlighting that the choice of -30 °C for shape fixing and 60 °C for shape recovery was made to facilitate rapid and complete crystallization and melting of the PEG domains, respectively, thereby ensuring fast shape fixation and recovery. The switching temperature of TPAE can be further tuned over a wide range by adjusting the ratio and type of soft and hard segments, which holds significant potential for various practical applications.
The Figs. 12. a-c illustrate the temperature memory effect of the TPAE, we have developed a force-induced temperature device. When the TPAE330 is bent and secured on a heating table set at 60°C, a small ball placed in proximity to the TPAE330 will cause the sample to visibly alter its shape after 25 s. At the 50s mark, the TPAE330 will propel the small ball away.
Successfully synthesized new temperature-responsive shape memory TPAEs with tunable blocks using a two-step melting method, incorporating a third monomer, BDO, to achieve the alcohol-acid equilibrium. The successful synthesis of TPAEs was confirmed by the presence of amide and ester bonds via FTIR and 1H NMR tests. Furthermore, the elastomers were found to be structurally stable at high temperatures, with decomposition temperatures exceeding 370.0°C as determined by TGA. The elastomers could be recycled and melted within a temperature range of 208.6°C to 370.0°C. Under room temperature conditions, the elastomers demonstrated an impressive elongation at a break of 592.4% while maintaining a tensile strength of 23.1 MPa. DMA was employed to investigate their thermo-mechanical properties, revealing two types of relaxation states around − 40°C and 50°C, which are similar to the results obtained from DSC. The DSC results indicated the presence of two melting temperatures, which corresponded to the soft and hard segments of the elastomer, respectively. Given that TPAEs are composed of weakly polarized PEG and strongly polarized PA512, they undergo microphase separation, which can be observed under SEM. Thus, the microphase separation of PEG and PA512 segments can be achieved to produce the microphase-separated structure of TPAEs This structure was prepared by maintaining the prepared TPAE at 60°C for 0.5 h, followed by bending and deformation of the strips. The strips were then fixed in a curved shape at -30°C for 0.5 h. The TPAE sample was heated to 60°C and then twisted, after which the helical shape was fixed by placing the strip in a -30°C environment for 5 min. Upon reheating to 60°C, the sample exhibited shape memory recovery, with TPAE230 demonstrating the most outstanding shape memory effect, achieving an average Rf of 91.2% and an Rr of 94.4%. The thermoplastic shape memory TPAEs, which possess good elastic recovery and thermal stability, have potential applications in sensors.
Acknowledgment
The authors thank the Taiyuan Institute of Technology for its help.
Author contribution Chengke Yuan and Jianyu Xue were responsible for writing the majority of the manuscript and conducting all experimental work. Jia Mi and Yu Wang contributed to portions of the text and assisted with revisions. Yingchun Li and Zhexenbek Toktarbay oversaw the experiments, managed the overall project, and participated in the manuscript revision process.
Funding: not applicable.
Competing interests: The authors declare no competing interests.
Ethical Approval: not applicable.
- B. Zhang, J.E. DeBartolo, J. Song, Shape Recovery with Concomitant Mechanical Strengthening of Amphiphilic Shape Memory Polymers in Warm Water, Acs Applied Materials & Interfaces 9(5) (2017) 4450-4456. https://10.1021/acsami.6b14167.
- Y.P. Hao, S.S. Liu, G.Y. Liu, A novel method for exploring the wear resistance characteristics of ethylene propylene diene monomers-polyamide elastomers with nano-graphite, Journal of Applied Polymer Science 141(14) (2024) 11. https://10.1002/app.55183.
- W.M. Peng, X. Tong, M.L. Zhang, X.J. Wang, G. Zhang, S.R. Long, J. Yang, Semiaromatic polyamide poly(hexamethylene terephthalamide)-co-polycaprolactam: Thermal and flame-retardant properties, Journal of Applied Polymer Science 135(29) (2018) 9. https://10.1002/app.46451.
- J. Wang, A. Reyna-Valencia, B.D. Favis, Continuity, morphology and surface resistivity in binary blends of poly(ether-block-amide) with polyethylene and polystyrene, European Polymer Journal 91 (2017) 197-211. https://10.1016/j.eurpolymj.2017.03.062.
- R. Casadei, M.G. Baschetti, M.J. Yoo, H.B. Park, L. Giorgini, Pebax(R) 2533/Graphene Oxide Nanocomposite Membranes for Carbon Capture, Membranes 10(8) (2020) 20. https://10.3390/membranes10080188.
- A.A. Jameh, T. Mohammadi, O. Bakhtiari, Preparation of PEBAX-1074/modified ZIF-8 nanoparticles mixed matrix membranes for CO2 removal from natural gas, Separation and Purification Technology 231 (2020) 20. https://10.1016/j.seppur.2019.115900.
- S.A. Mohammed, A.M. Nasir, F. Aziz, G. Kumar, W. Sallehhudin, J. Jaafar, W.J. Lau, N. Yusof, W.N.W. Salleh, A.F. Ismail, CO2/N2 selectivity enhancement of PEBAX MH 1657/Aminated partially reduced graphene oxide mixed matrix composite membrane, Separation and Purification Technology 223 (2019) 142-153. https://10.1016/j.seppur.2019.04.061.
- S. Todros, A.N. Natali, M. Piga, G.A. Giffin, G. Pace, V. Di Noto, Interplay between chemical structure and ageing on mechanical and electric relaxations in poly(ether-block-amide)s, Polymer Degradation and Stability 98(6) (2013) 1126-1137. https://10.1016/j.polymdegradstab.2013.03.014.
- M.R. Barzegari, N. Hossieny, D. Jahani, C.B. Park, Characterization of hard-segment crystalline phase of poly(ether-block-amide) (PEBAX®) thermoplastic elastomers in the presence of supercritical CO2 and its impact on foams, Polymer 114 (2017) 15-27. https://10.1016/j.polymer.2017.02.088.
- X. Tong, W.M. Peng, M.L. Zhang, X.J. Wang, G. Zhang, S.R. Long, J. Yang, A new class of poly(ether-block-amide)s based on semi-aromatic polyamide: synthesis, characterization and structure-property relations, Polymer International 70(2) (2021) 230-241. https://10.1002/pi.6119.
- P. Fu, J.H. Zhang, Y.C. Zhang, W. Zhao, X.M. Zhang, G. Shi, Y.J. He, Z. Cui, X.C. Pang, M.Y. Liu, Shape Memory Thermoplastic Polyamide Elastomer Based on PA1212, Macromolecular Materials and Engineering 308(4) (2023) 12. https://10.1002/mame.202200547.
- C.W. Yi, Z.H. Peng, H.P. Wang, M. Li, C.S. Wang, Synthesis and characteristics of thermoplastic elastomer based on polyamide-6, Polymer International 60(12) (2011) 1728-1736. https://10.1002/pi.3140.
- X.B. Fu, T. Zhang, J.C. Yang, G. Zhang, M.L. Zhang, X.J. Wang, J. Yang, Structures and properties of newly synthesized semi-aromatic polyamide thermoplastic elastomers, Polymer Chemistry 13(34) (2022) 4980-4991. https://10.1039/d2py00541g.
- J. Jiang, Q.Y. Tang, X. Pan, J.J. Li, L. Zhao, Z.H. Xi, W.K. Yuan, Facile Synthesis of Thermoplastic Polyamide Elastomers Based on Amorphous Polyetheramine with Damping Performance, Polymers 13(16) (2021) 19. https://10.3390/polym13162645.
- P. Lu, Z.Y. Zhao, B.R. Xu, Y.M. Li, C. Deng, Y.Z. Wang, A novel inherently flame-retardant thermoplastic polyamide elastomer, Chemical Engineering Journal 379 (2020) 12. https://10.1016/j.cej.2019.122278.
- Y. Shibasaki, T. Mori, A. Fujimori, M. Jikei, H. Sawada, Y. Oishi, Poly(amide-ether) Thermoplastic Elastomers Based on Monodisperse Aromatic Amide Hard Segments as Shape-Memory and Moisture-Responsive Materials, Macromolecules 51(23) (2018) 9430-9441. https://10.1021/acs.macromol.8b01817.
- M. Peyravi, M.A. Ardestani, A.A. Babaluo, M.K.R. Aghjeh, S.R. Pishghadam, E. Jannatdoust, SYNTHESIS AND CHARACTERIZATION OF NANOSTRUCTURED-SEGMENTED BLOCK COPOLYETHERAMIDE BASED ON NYLON6 AND POLY(ETHYLENE OXIDE), Chinese Journal of Polymer Science 28(4) (2010) 597-605. https://10.1007/s10118-010-9083-0.
- J. Jiang, Q.Y. Tang, X. Pan, Z.H. Xi, L. Zhao, W.K. Yuan, Structure and Morphology of Thermoplastic Polyamide Elastomer Based on Long-Chain Polyamide 1212 and Renewable Poly(trimethylene glycol), Industrial & Engineering Chemistry Research 59(39) (2020) 17502-17512. https://10.1021/acs.iecr.0c01334.
- J.M. Robertson, H.B. Nejad, P.T. Mather, Dual-Spun Shape Memory Elastomeric Composites, Acs Macro Letters 4(4) (2015) 436-440. https://10.1021/acsmacrolett.5b00106.
- Z. Sang, K. Ke, I. Manas-Zloczower, Effect of carbon nanotube morphology on properties in thermoplastic elastomer composites for strain sensors, Composites Part a-Applied Science and Manufacturing 121 (2019) 207-212. https://10.1016/j.compositesa.2019.03.007.
- M. Wu, L. Yuan, F. Jiang, Y.Q. Zhang, Y.R. He, Y.Z. You, C.B. Tang, Z.K. Wang, Strong Autonomic Self-Healing Biobased Polyamide Elastomers, Chemistry of Materials 32(19) (2020) 8325-8332. https://10.1021/acs.chemmater.0c02169.
- W.B. Kong, K. Hu, X.W. Fu, D.Y. Guo, J.X. Lei, Preparation and Characterization of Thermoplastic Elastomer Based on Amino-terminated Polyamide-6 and Diisocyanate-terminated Polytetramethylene Glycol, Polymer-Plastics Technology and Engineering 55(1) (2016) 1-8. https://10.1080/03602559.2015.1050510.
- S.M. Peng, L. Peng, C.W. Yi, W. Zhang, X. Wang, A novel synthetic strategy for preparing semi-aromatic components modified polyamide 6 polymer, Journal of Polymer Science Part a-Polymer Chemistry 56(9) (2018) 959-967. https://10.1002/pola.28983.
- Y. Nurhamiyah, G. Irvine, E. Themistou, B.Q. Chen, Novel Biobased Polyamide Thermoplastic Elastomer with Medium Hardness, Macromolecular Chemistry and Physics 222(21) (2021) 7. https://10.1002/macp.202100218.
- J. Chen, C.H. Gong, C. Yang, C.W. Yi, Flexible preparation of polyamide-6 based thermoplastic elastomers via amide exchange, Journal of Materials Science 56(20) (2021) 12018-12029. https://10.1007/s10853-021-06057-z.
- M. Peyravi, A.A. Babaluo, M.A. Ardestani, M.K.R. Aghjeh, S.R. Pishghadam, P. Hadi, Study on the Synthesis of Poly(ether-block-amide) Copolymer Based on Nylon6 and Poly(ethylene oxide) with Various Block Lengths, Journal of Applied Polymer Science 118(2) (2010) 1211-1218. https://10.1002/app.32358.
- R.C. Yuan, S. Fan, D.Q. Wu, X.L. Wang, J.Y. Yu, L.J. Chen, F.X. Li, Facile synthesis of polyamide 6 (PA6)-based thermoplastic elastomers with a well-defined microphase separation structure by melt polymerization, Polymer Chemistry 9(11) (2018) 1327-1336. https://10.1039/c8py00068a.
- Y.I. Odarchenko, N.J. Sijbrandi, M. Rosenthal, A.J. Kimenai, E.P.C. Mes, R. Broos, G. Bar, P.J. Dijkstra, J. Feijen, D.A. Ivanov, Structure formation and hydrogen bonding in all-aliphatic segmented copolymers with uniform hard segments, Acta Biomaterialia 9(4) (2013) 6143-6149. https://10.1016/j.actbio.2012.09.038.
- R.L. Zhu, Z.J. Pu, H.B. Hou, X.Y. Li, X. Wang, D.Y. Yu, P. Zheng, F. Wu, J.C. Zhong, Effect of copolymerization on the thermal characteristics and behavior of crystallization of biobased semi-aromatic PA10T/10I, Journal of Applied Polymer Science 140(22) (2023) 8. https://10.1002/app.53903.
- Y. Nurhamiyah, A. Amir, M. Finnegan, E. Themistou, M. Edirisinghe, B.Q. Chen, Wholly Biobased, Highly Stretchable, Hydrophobic, and Self-healing Thermoplastic Elastomer, Acs Applied Materials & Interfaces 13(5) (2021) 6720-6730. https://10.1021/acsami.0c23155.
- W.Z. Wang, X.W. Wang, R.X. Li, B.Y. Liu, E.G. Wang, Y.H. Zhang, Environment-Friendly Synthesis of Long Chain Semiaromatic Polyamides with High Heat Resistance, Journal of Applied Polymer Science 114(4) (2009) 2036-2042. https://10.1002/app.30774.
- Y. Feng, Y.C. Li, X.M. Ye, Z.M. Li, W.S. Wang, T. Liu, I.H. El Azab, G.A.M. Mersal, M.M. Ibrahim, Z.M. El-Bahy, M.N. Huang, Z.H. Guo, Synthesis and characterization of 2,5-furandicarboxylic acid poly(butanediol sebacate-butanediol) terephthalate (PBSeT) segment copolyesters with excellent water vapor barrier and good mechanical properties, Journal of Materials Science 57(24) (2022) 10997-11012. https://10.1007/s10853-022-07269-7.
- G.P. Baeza, A. Sharma, A. Louhichi, L. Imperiali, W.P.J. Appel, C.F.C. Fitié, M.P. Lettinga, E. Van Ruymbeke, D. Vlassopoulos, Multiscale organization of thermoplastic elastomers with varying content of hard segments, Polymer 107 (2016) 89-101. https://10.1016/j.polymer.2016.11.010.
- X.C. Xun, X. Zhao, Q. Li, B. Zhao, T. Ouyang, Z. Zhang, Z. Kang, Q.L. Liao, Y. Zhang, Tough and Degradable Self-Healing Elastomer from Synergistic Soft-Hard Segments Design for Biomechano-Robust Artificial Skin, Acs Nano 15(12) (2021) 20656-20665. https://10.1021/acsnano.1c09732.
- D. Husken, T. Visser, M. Wessling, R.J. Gaymans, CO2 permeation properties of poly(ethylene oxide)-based segmented block copolymers, Journal of Membrane Science 346(1) (2010) 194-201. https://10.1016/j.memsci.2009.09.034.
- H.S. Guo, Y. Han, W.Q. Zhao, J. Yang, L. Zhang, Universally autonomous self-healing elastomer with high stretchability, Nature Communications 11(1) (2020) 9. https://10.1038/s41467-020-15949-8.
- H. Wang, J.H. Xu, D.M. Xing, X.S. Du, H.B. Wang, Z.L. Du, X. Cheng, Healable and reprocessable silica/poly(oxime-urethane) composite elastomer with high mechanical robustness and exceptional damage-tolerant capacity, Journal of Applied Polymer Science 139(26) (2022) 12. https://10.1002/app.52472.
- P. Zhu, X. Dong, M.M. Huang, L.L. Wang, S.X. Qi, D.J. Wang, Microstructural Evolution Underlying the Ternary Stages of the Elastic Behaviors for Poly(Ether-b-Amide) Copolymer Elastomers, Journal of Polymer Science Part B-Polymer Physics 56(11) (2018) 855-864. https://10.1002/polb.24600.
- Y.T. Zhang, Y.J. Shen, J.W. Hou, Y.M. Zhang, W. Fam, J.D. Liu, T.D. Bennett, V. Chen, Ultraselective Pebax Membranes Enabled by Templated Microphase Separation, Acs Applied Materials & Interfaces 10(23) (2018) 20006-20013. https://10.1021/acsami.8b03787.
- Y.L. Ding, C.X. Zhang, B.B. Shi, Y.Y. Wang, P.F. Tang, C. Liu, J.J. Fan, Z.K. Wang, F. Jiang, Sustainable ultra-strong polyesteramide elastomers with rapid degradation and high resilience, European Polymer Journal 210 (2024) 8. https://10.1016/j.eurpolymj.2024.112901.
- Q.H. Meng, J.L. Hu, Influence of heat treatment on the properties of shape memory fibers. I. Crystallinity, hydrogen bonding, and shape memory effect, Journal of Applied Polymer Science 109(4) (2008) 2616-2623. https://10.1002/app.28363.
- Y. Song, Y. Liu, T. Qi, G.L. Li, Towards Dynamic but Supertough Healable Polymers through Biomimetic Hierarchical Hydrogen-Bonding Interactions, Angewandte Chemie-International Edition 57(42) (2018) 13838-13842. https://10.1002/anie.201807622.
- S. Pisharath, X. Hu, S.C. Wong, Rheology-morphology relationships in nylon-LCP hybrid composites, Composites Science and Technology 66(15) (2006) 2971-2979. https://10.1016/j.compscitech.2006.02.018.
- R. Shibata, T. Ishihara, T. Tsukamoto, Y. Oishi, A. Fujimori, Y. Shibasaki, Synthesis of tetraazacalix 2 arene 2 triazine-containing poly (dimethylsiloxane) with elastic property induced by pinning effect of the calixarene ring, European Polymer Journal 162 (2022) 11. https://10.1016/j.eurpolymj.2021.110890.
- H.B. Lu, W.M. Huang, Y.T. Yao, Review of chemo-responsive shape change/memory polymers, Pigment & Resin Technology 42(4) (2013) 237-246. https://10.1108/prt-11-2012-0079.
- W. Zhang, L. Chen, Y. Zhang, Surprising shape-memory effect of polylactide resulted from toughening by polyamide elastomer, Polymer 50(5) (2009) 1311-1315. https://10.1016/j.polymer.2009.01.032.
- W.J. Lu, P. Zhu, Y. Zhao, D.J. Wang, X. Dong, A Disulfide-Based Poly(ether-b-amide) Copolymer with Rapid Self-healing Ability under Moderate Conditions, Chinese Journal of Chemistry (2024) 8. https://10.1002/cjoc.202300603.
Scheme 1 is available in the Supplementary Files section.
No competing interests reported.
- Scheme1.png
Scheme 1. The synthesis route of PrePA512 and TPAE.
{kind=link}