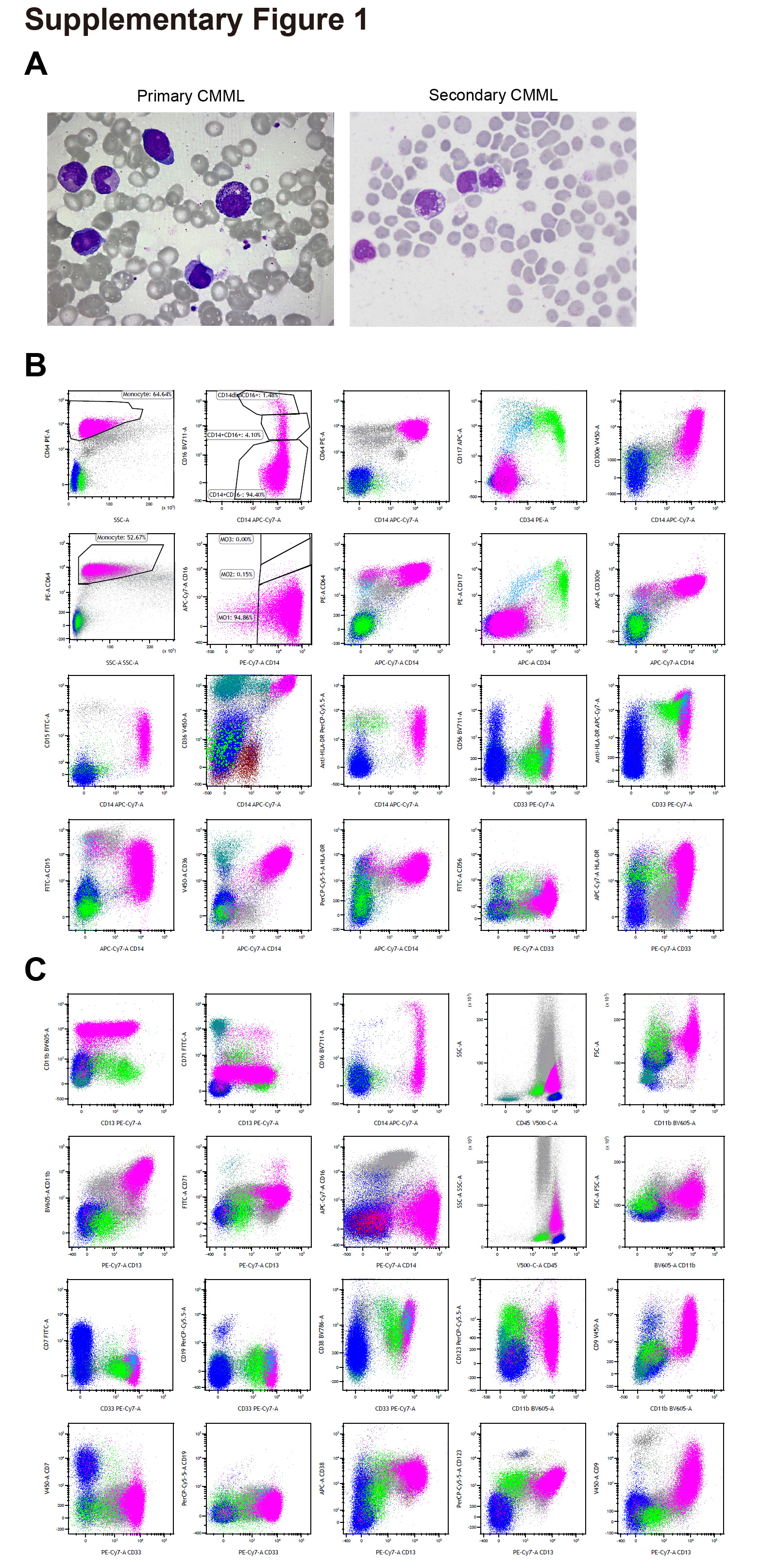
3.1. Morphological and immunophenotypic differences between primary and secondary CMML
To identify the potential heterogeneities of secondary CMML and primary CMML, we analyzed the morphology and immunophenotypes of secondary CMML and primary CMML. Our results indicated that secondary CMML present distinct morphology and immunophenotypes compared with primary CMML (Supplementary Fig. 1A, B and C, Supplementary Table 1). In conclusion, there may be important differences between primary CMML and secondary CMML, as shown by the results of the morphological and flowcytometry immunophenotyping analyses.
3.2. Bone marrow immune microenvironment features of primary CMML, and secondary CMML before treatment
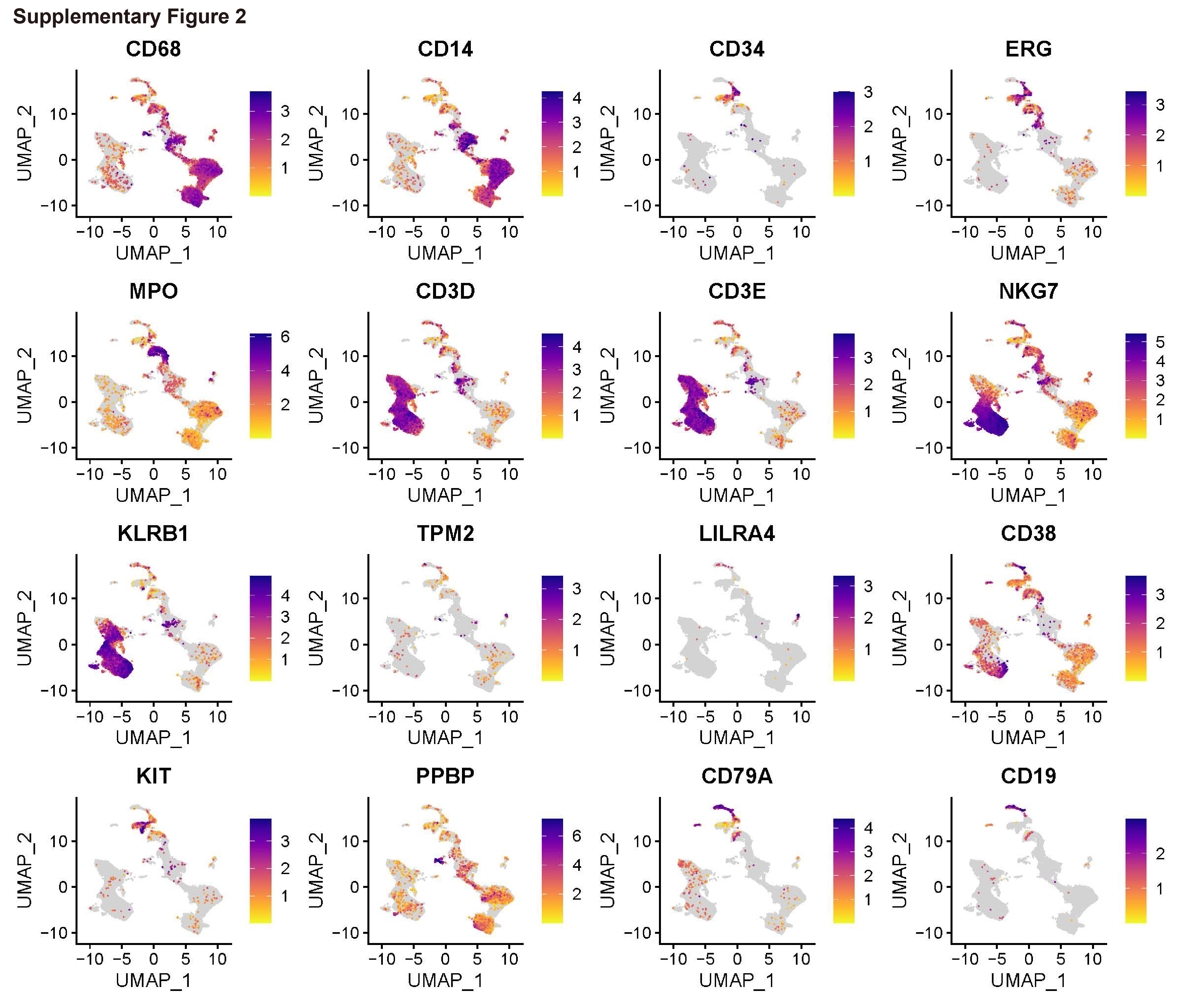
To generate a single-cell transcriptome landscape of BM CD45+ cells in primary CMML, secondary CMML, and HCs, we collected fresh BM samples from patients with primary CMML and secondary CMML. Subsequently, ScRNA-seq was performed on the CD45+ BM cells (Fig. 1A). The ScRNA-seq of BM samples of HCs were obtained from You et al. 15 After stringent quality control, a total of 37,011 cells were obtained from four BM CD45+ samples and further classified into seven cell lineages (Fig. 1B and C, Supplementary Table 2). Based on the expression of canonical marker genes (Fig. 1D and Supplementary Fig. 2), we defined them as monocytes/macrophages (CD68, CD14), HSCs/HSPCs (CD34, ERG), granulocytes (MPO), T/NK cells (CD3D, CD3E, NKG7, KLRB1), DCs (TMP2, LILRA4), GMPs (CD38, KIT, PPBP), and B cells (CD79A, CD19). Significantly, patients with secondary CMML had the lowest ratio of T/NK cells and the highest ratio of monocytes/macrophages (Fig. 1E). This finding was also supported by an increased expression of monocytes/macrophages-related genes such as LYZ, CD14, MAFB, and MNDA and a decreased expression of T/NK cell differentiation-related genes CD52, KLRB1, TRAC, NKG7, GZMA, GZMB, and GNLY (Fig. 1F). Patients with secondary CMML exhibited heightened infection, inflammation, and phagocytosis pathways (Fig. 1G), alongside reduced T and B cell receptor signaling (Fig. 1H). This suggests that secondary CMML development may be linked to impaired T/B cell immune responses, leading to secondary infections and subsequent activation of monocytes/macrophages, causing systemic inflammation and contributing to CMML progression.
3.3. Increased infection-mediated phagocytosis, chemotaxis and immune responses of monocytes/macrophages in secondary CMML
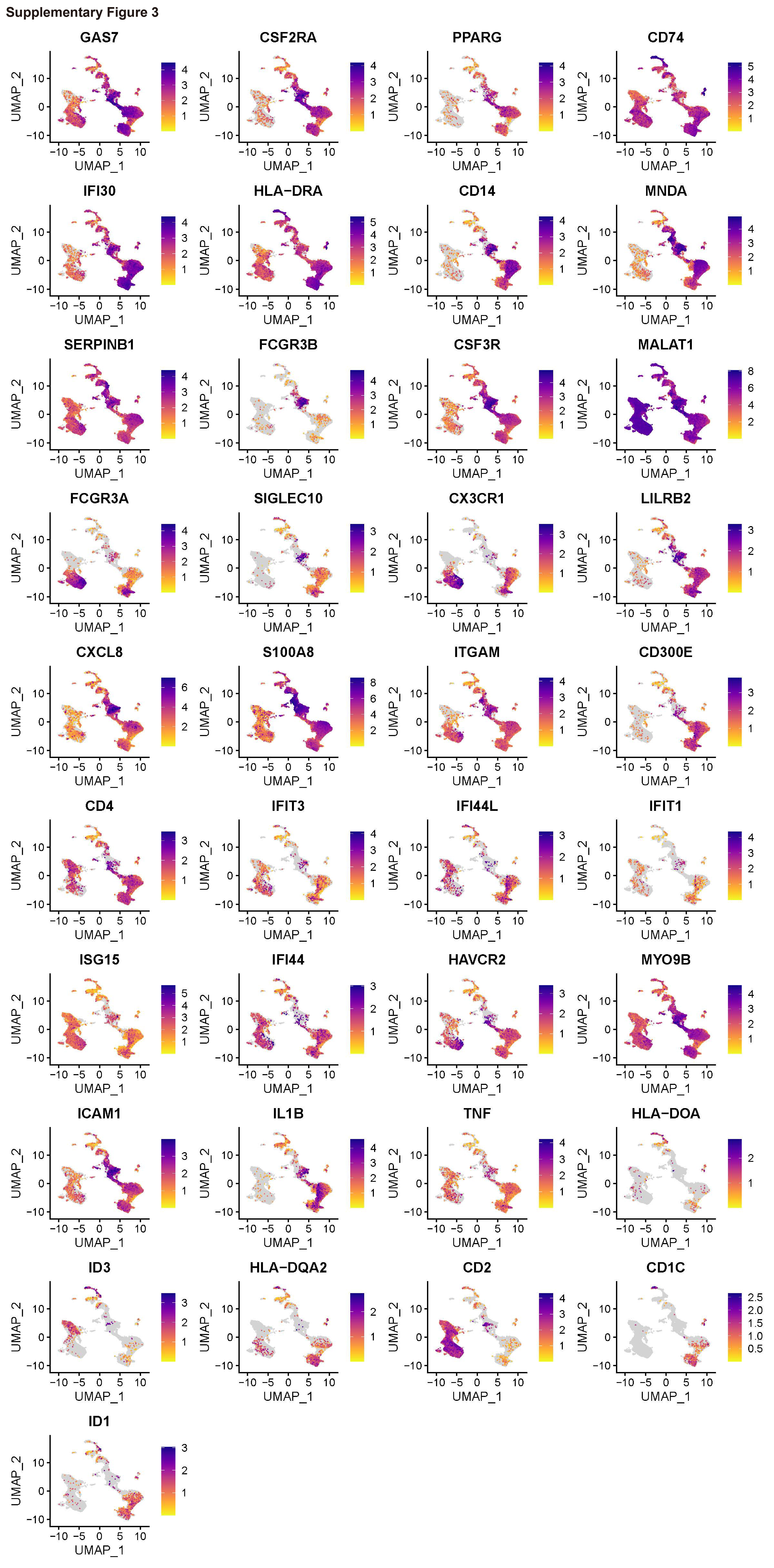
We next divided monocytes/macrophages into 11 subpopulations based on marker genes (Figs. 2A, B, and C, Supplementary Table 3). Cluster 0 showed a higher level of expression of GAS7, CSF2RA, and PPARG; Cluster 1 was characterized by higher expression levels of CD74, IFI30, and HLA-DRA; Cluster 2 was reported to express high levels of CD14, MNDA, and SERPINB1; Cluster 3 expressed higher levels of FCGR3B, CSF3R, and MALAT1; Cluster 4 expressed high levels of FCGR3A, SIGLEC10, CX3CR1, and LILRB2; Cluster 5 expressed higher levels of CXCL8 and S100A8; Cluster 6 demonstrated higher levels of expression of ITGAM, CD300E, and CD4; Cluster 7 showed a higher level of expression of IFIT3, IFI44L, IFIT1, ISG15, and IFI44; Cluster 8 showed high levels of expression of HAVCR2 and MYO9B; Cluster 9 expressed high levels of ICAM1, IL1B, and TNF; and Cluster 10 was reported to express high levels of HLA-DOA, ID3, HLA-DQA2, CD2, CD1C, and ID1 (Fig. 2D, Supplementary Fig. 3). Interestingly, the secondary CMML patient demonstrated an increased percentage of Clusters 1, 2, 3, 7, and 10 but a decreased percentage of Clusters 0, 4, 5,6, 8, and 9 (Fig. 2E). The upregulated KEGG pathways enriched for DEGs in Clusters 1, 2, 3, 7, and 10 included antigen processing and presentation, coronavirus disease (COVID-19), oxidative phosphorylation, chemokine signaling pathway, a neurotrophin signaling pathway, endocytosis, Fc gamma R-mediated phagocytosis, and influenza A. However, Cluster 0, which has upregulated KEGG pathways, including ubiquitin mediated proteolysis, thyroid hormone signaling pathway, and MAPK signaling pathway, was only found in primary CMML. The upregulated KEGG pathway in Clusters 4, 5, 6, 8, and 9 included Th1 and Th2 cell differentiation, a T cell receptor signaling pathway, PD-L1 expression and a PD-1 checkpoint pathway in cancer, an Epstein–Barr virus infection, lipids and atherosclerosis, an NF-kappa B signaling pathway, and a Kaposi sarcoma-associated herpesvirus infection (Fig. 2F). Collectively, these results indicated that monocytes/macrophages in secondary CMML before treatment correlated with an infection-mediated phagocytosis, chemotaxis and immune responses microenvironment, which may contribute to the development of CMML.
3.4. Transcriptomic differences of the monocytes/macrophages between primary CMML and secondary CMML
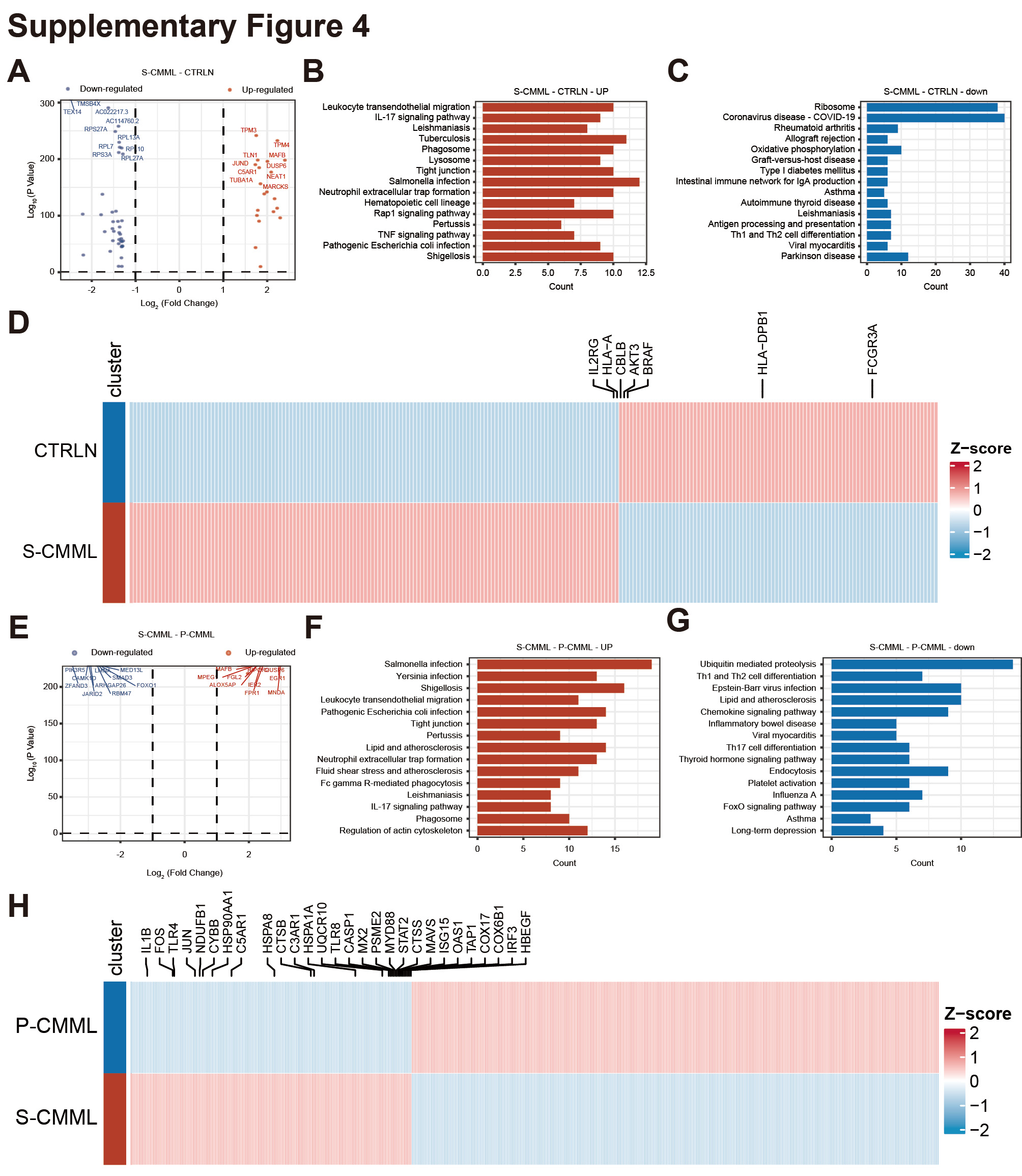
We next elucidated the transcriptomic differences between the monocytes/macrophages of primary CMML and secondary CMML. In total, the overall DEG analysis showed that 1673 genes were upregulated, whereas 582 were downregulated in secondary CMML compared to HCs (Supplementary Fig. 4A, Supplementary Tables 4 and 5). The KEGG pathway enrichment of upregulated genes in secondary CMML included salmonella infection, pathogenic Escherichia coli infection, human cytomegalovirus infection, yersinia infection, tuberculosis, viral carcinogenesis, phagosomes, lysosomes, and Fc gamma R-mediated phagocytosis (Supplementary Fig. 4B). In contrast, the KEGG pathway enrichment of downregulated genes included coronavirus disease (COVID-19), oxidative phosphorylation, antigen processing and presentation, and viral myocarditis (Supplementary Fig. 4C). Specifically, the expression levels of genes encoding proteins known to be important for the KEGG pathways were significantly higher in secondary CMML (Supplementary Fig. 4D). Subsequently, an analysis of the DEGs of patients with secondary and primary CMML showed that 1588 genes were upregulated, whereas 1834 were downregulated in secondary CMML compared to primary CMML (Supplementary Fig. 4E, Supplementary Tables 6 and 7). The KEGG pathway enrichment of upregulated genes in secondary CMML patient included salmonella infection, phagosomes, coronavirus disease (COVID − 19), pathogenic Escherichia coli infection, oxidative phosphorylation, and yersinia infection (Supplementary Fig. 4F). In contrast, the KEGG pathway enrichment of downregulated genes included chronic myeloid leukemia, T cell receptor signaling, a chemokine signaling pathway, an Epstein–Barr virus infection, and a Kaposi sarcoma-associated herpesvirus infection (Supplementary Fig. 4G). Specifically, the expression levels of genes encoding proteins known to be important for the KEGG pathways were significantly higher in secondary CMML (Supplementary Fig. 4H). These findings collectively suggest that increased infection in secondary CMML may correlate with the occurrence of this disease.
3.5. Decreased cytotoxicity of T/NK cells in patients with secondary CMML before treatment
We further identified subclusters in the T/NK cells. A total of 16 subpopulations were identified based on marker genes (Figs. 3A, B, C, and D, Supplementary Table 8). Based on gene expression profiling, isolated cells from the BM were then categorized into 11 main subclusters, including three subtypes of CD4+Naïve T (Tn, Clusters 0, 5, and 8), four subtypes of early effector memory T cell (early Tem, Clusters 1,2,3 and 9), one subtype of Temra (Cluster 6), one type of NK-like cells (Cluster 7), one subtype of Tc17 (Cluster 10), one subtype of γδ-T cells (Cluster 11), one subtype of Treg (Cluster 13), one subtype of Tcm (Cluster 14), and one subtype of proliferating T cells (Cluster 15) (Fig. 3E, Supplementary Table 8). Cluster 13 included T cell markers and myeloid cell markers. Interestingly, the percentages of Clusters 0, 1, and 15 were increased in secondary CMML (Fig. 3F). After merging the same cell types, we found that the percentages of early Tem, proliferating T cells, Tcm, and Treg increased, while the percentage of γδ-T cells, NK-like cells, NKT cells, and Tn cells decreased, which may also contribute to the occurrence and development of secondary CMML (Fig. 3G).
3.6. Transcriptomic differences of T/NK cells between the primary CMML and secondary CMML
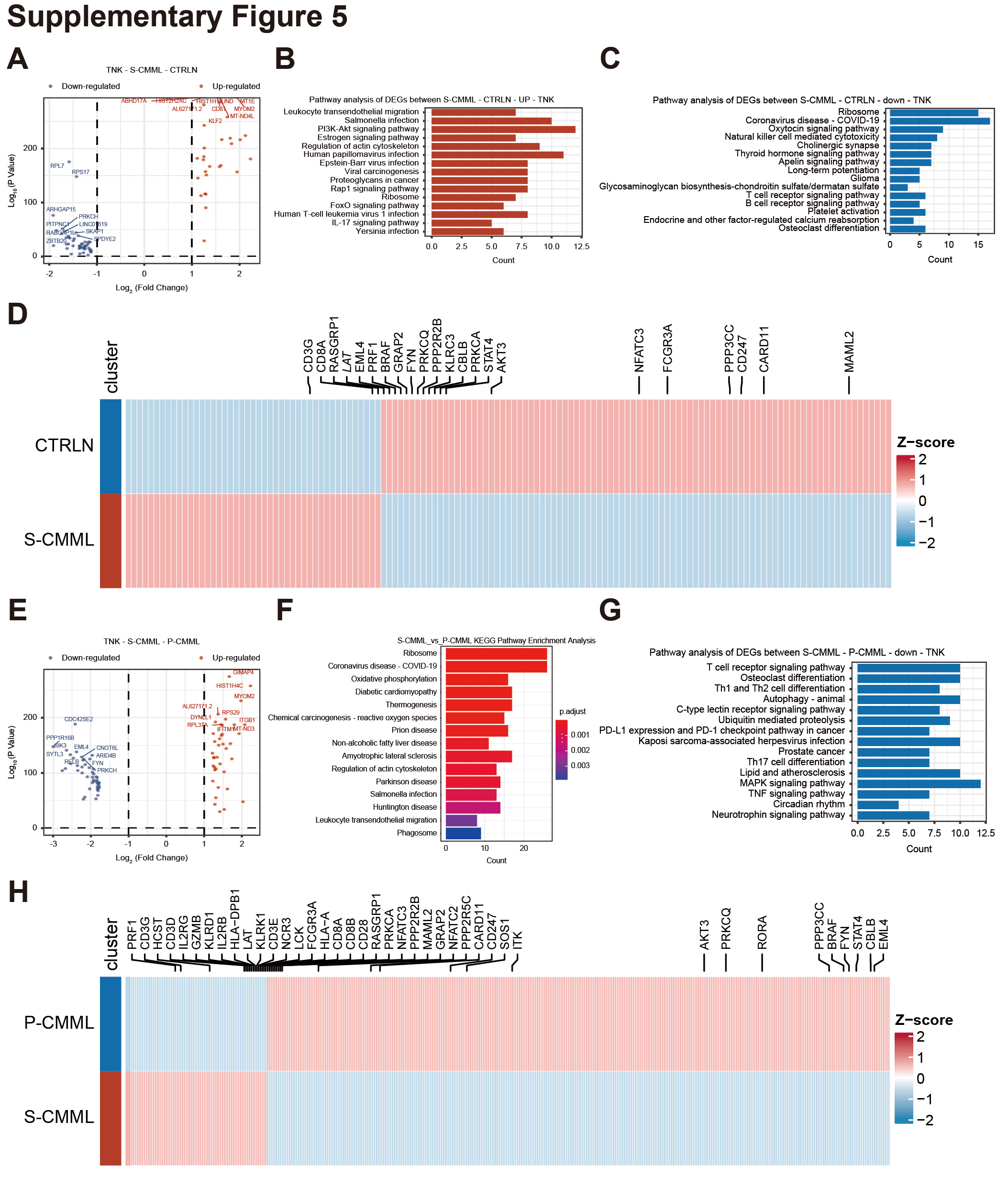
We also analyzed transcriptional differences in T/NK cells of patients with primary CMML and secondary CMML. A DEG analysis indicated that 589 genes were upregulated, whereas 694 were downregulated in secondary CMML, compared to HCs (Supplementary Fig. 5A, Supplementary Tables 9 and 10). The KEGG pathway enrichment of upregulated genes in patients with secondary CMML included a salmonella infection, a human papillomavirus infection, an Epstein–Barr virus infection, viral carcinogenesis, a human T-cell leukemia virus 1 infection, and a Yersinia infection (Supplementary Fig. 5B). In contrast, the KEGG pathway enrichment of downregulated genes included natural killer cell-mediated cytotoxicity, and a T cell receptor signaling pathway (Supplementary Fig. 5C). Specifically, the expression levels of genes encoding proteins known to be important for the KEGG pathways were significantly higher in secondary CMML (Supplementary Fig. 5D). Subsequently, an analysis of the DEGs of patients with secondary CMML and with primary CMML showed that 1314 genes were upregulated, whereas 969 were downregulated in secondary CMML compared to primary CMML (Supplementary Fig. 5E, Supplementary Tables 11 and 12). The KEGG pathway enrichment of upregulated genes in secondary CMML included salmonella infection, phagosomes, oxidative phosphorylation, coronavirus disease (COVID-19), and leukocyte transendothelial migration (Supplementary Fig. 5F). In contrast, the KEGG pathway enrichment of downregulated genes included T cell receptor signaling, osteoclast differentiation, Th1 and Th2 cell differentiation, and Th17 cell differentiation (Supplementary Fig. 5G). Specifically, the expression levels of genes encoding proteins known to be important for the KEGG pathways were significantly higher in secondary CMML (Supplementary Fig. 5H). Taken together, secondary CMML patients have reduced tumor killing activity of T/NK cells in the BM microenvironment.
3.7. The potential indirect mechanisms of secondary CMML via cell–cell communication analysis
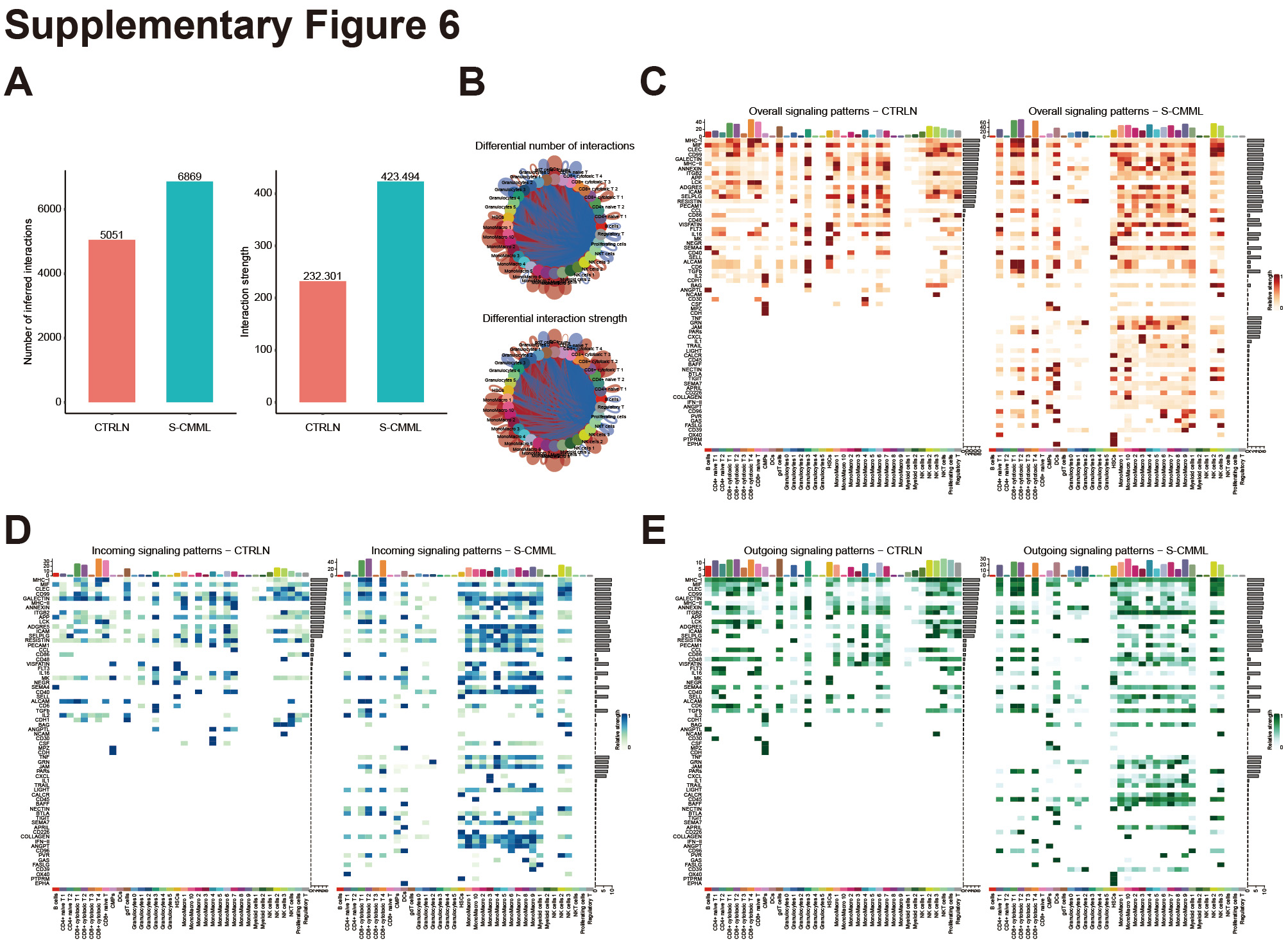
To study cell–cell communication in secondary CMML, we compared its signaling pathways with those of primary CMML. Figures 4A and B show that secondary CMML has fewer but stronger interactions than primary CMML. We then analyzed the signaling pathways in each cell population of both primary and secondary CMML. Figure 4C indicates that the overall signaling pathways in secondary CMML included IL16, CXCL, CCL, TNF, PARS, IL1, TRAIL, CD40, BTLA, CSF, GAS, IL2, TIGIT, and others. The incoming signaling patterns of secondary CMML included TIGIT, IL16, CXCL, IL1, TRAIL, CSF, GAS, IL2, and others so on (Fig. 4D). The outgoing signaling patterns of secondary CMML included TIGIT, IL16, CXCL, IL1, TRAIL, PVR, GAS, IL2, and others (Fig. 4E). We then performed a comparative analysis of secondary CMML and HC cell communication differences, and the results are consistent with the above (Supplementary Figs. 6A–E). Collectively, our results suggest that the monocytes/macrophages from secondary CMML are associated with inflammatory cytokines, chemokines, and immunosuppression, which may induce T/NK dysfunction.
3.8. The remodeled BM immune cells in secondary CMML post Azacitidine plus Venecla treatment
Azacitidine in combination with Venetoclax is often used in the treatment of CMML. 16–18 To understand the effects of Azacitidine plus Venecla on secondary CMML patients, we analyzed immune cell cluster changes pre- and post-treatment. CD45+ cells were categorized into T/NK cells, monocytes/macrophages, B cells, CMPs, granulocytes, DCs, and HSCs (Fig. 5A). Figures 5B and 5C display cells from both the before-treatment and after-treatment groups. Post-treatment, the patient showed a higher percentage of T/NK cells and a lower percentage of monocytes/macrophages and granulocytes (Fig. 5D). In line with this result, most of the top 10 DEGs before treatment are related to monocytes/macrophages (e.g., VCAN, LYZ, DUSP6), while most of the top 10 DEGs after treatment are related to T/NK cells (e.g., IL32, IL7R, CCL5, Fig. 5E). The KEGG pathway enrichment of upregulated genes in the after-treatment group included antigen processing and presentation, natural killer cell-mediated cytotoxicity, and a T cell receptor signaling pathway (Fig. 5F). In contrast, the KEGG pathway enrichment of downregulated genes in the after-treatment group included lysosomes, phagosomes, and Fc gamma R-mediated phagocytosis (Fig. 5G). Overall, our results suggest that Azacitidine plus Venecla treatment remodeled the immune microenvironment of patients with secondary CMML towards an anti-tumor microenvironment.
3.9. The effect on monocytes/macrophages of Azacitidine plus Venecla treatment in secondary CMML
We next reclustered monocytes/macrophages and divided them into seven subpopulations (Fig. 6A). The monocytes/macrophages derived from the before-treatment or after-treatment group are shown in Figs. 6B and C. Interestingly, after treatment, the patient demonstrated an increased percentage of Clusters 0 and 6 but a decreased percentage of Clusters 1–5 (Fig. 6D). We then analyzed the top six DEGs of these seven subclusters. The marker genes of Cluster 0 are FCGR3B, MMP9, IFITM2, MME, CMTM2, and LITAF, and the marker genes of Cluster 6 are HLA-DQA1, HLA-DQB1, HLA-DPB1, HLA-DPA1, HLA-DRA, and CD74 (Fig. 6E). The KEGG pathway enrichment of upregulated genes in the after-treatment group included antigen processing and presentation, an IL17 signaling pathway, and cytokine–cytokine receptor interaction, which may contribute to the regulation of the T/NK cell cytotoxicity effect (Fig. 6F). In contrast, the KEGG pathway enrichment of downregulated genes in the after-treatment group included lysosomes, coronavirus disease (COVID-19), and efferocytosis (Fig. 6G). Collectively, these findings suggest that Azacitidine plus Venecla treatment for secondary CMML may improve the patient's BM immune microenvironment from a pro-tumor progression microenvironment to an anti-tumor immune microenvironment.
3.10. Sub-clustering of T/NK lineage in secondary CMML before and after Azacitidine plus Venecla treatment
We further identified the changes in T/NK cells post Azacitidine plus Venecla treatment. We divided the T/NK cells from the before-treatment group or the after-treatment group into 13 subpopulations (Fig. 7A). Cluster 0 was identified as NK-1 cells based on the expression of NKG7, GZMA, GZMB, and GZMH; Cluster 1 was identified as naïve T cells based on the expression of LTB, IL7R, EEF1A1, and TPT1; Cluster 2 was identified as proliferating T cells based on the expression of MKI67, PCLAF, STMN1, HMGB2, and TUBA1B; and Clusters 3 and 7 were identified as double cells based on non-T/NK markers expression (CSF3R, MNCA, NAMPT, S100A8, S100A9, S100A12, CST3, IFI30, LYZ, AIF1, LST1, and CTSS). Cluster 4 was identified as γδ-T cells based on the expression of TRDC, TRGC1, KLRC2, CCL4, and GNLY. Cluster 6 was identified as CD8+ cytotoxicity T cells based on the expression of CD8B, CCR7, RPS6, RPS5, and LEF1. Cluster 8 was identified as NEAT1-T cells based on the expression of NEAT1, RNF213, and MALAT1. Cluster 9 was identified as CCR9+T cells based on the expression of CCR9, CD59 and TRBC2 (Fig. 7B and Supplementary Table 13). The T/NK cells derived from the before-treatment group or after-treatment group are shown in Fig. 7C. Interestingly, the percentage of Naïve T 3, NK1, NK2, and proliferation T increased in the after-treatment group (Fig. 7D). The KEGG pathway enrichment of upregulated genes in the after-treatment group included a NOD-like receptor signaling pathway, natural killer cell-mediated cytotoxicity, a chemokine signaling pathway, cytokine-cytokine receptor interaction, a Toll-like receptor signaling pathway, and a T cell receptor signaling pathway, which demonstrates the anti-tumor features induced by Azacitidine plus Venecla treatment (Fig. 7E). After treatment, downregulated genes were enriched in pathways related to various infections and viral carcinogenesis, likely due to decreased monocytes/macrophages and increased T/NK cells (Fig. 7F). Overall, myeloid cells showed significant transcriptional changes post-treatment with Azacitidine plus Venecla, potentially affecting T/NK cell function in multiple ways.
3.11. Cell–cell communication analysis demonstrated that monocytes/macrophages are key players in secondary CMML post treatment with Azacitidine plus Venecla
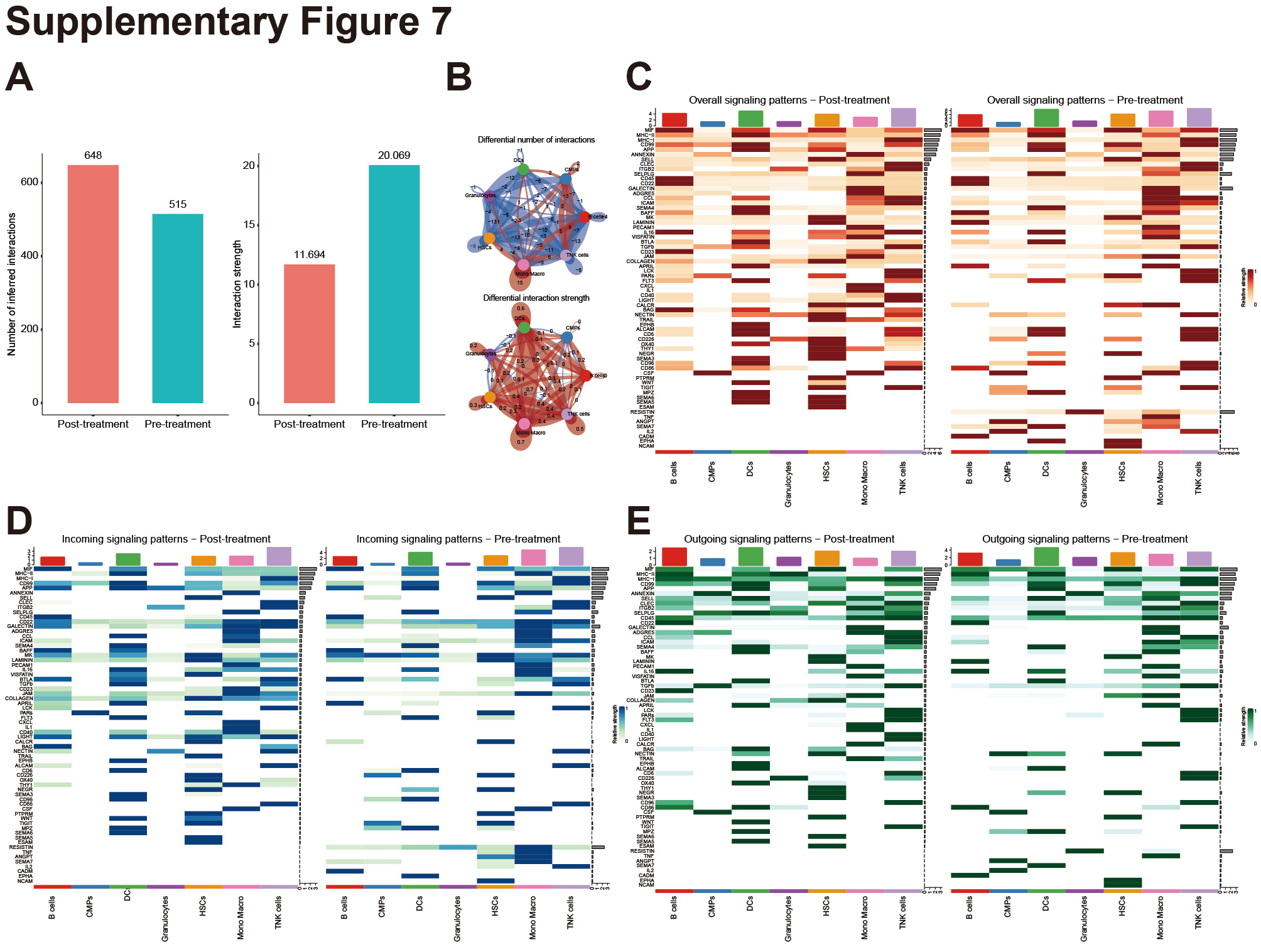
Finally, we performed a cell–cell communication analysis on cells from a patient with secondary CMML post treatment with Azacitidine plus Venecla using CellChat software. Supplementary Figs. 7A and B demonstrate that the number of inferred interactions increased, while the interaction strength decreased in the secondary CMML group post treatment with Azacitidine plus Venecla compared to the number before treatment. Thereafter, we analyzed the overall signaling pathways of each cell population, both after and before treatment. Supplementary Fig. 7C indicates that the overall signaling pathways that correlate to monocytes/macrophages in the after-treatment group included apoptosis-related genes, THY1, 19 TRAIL, 20 and APRIL, 21 which correlated with decreased numbers of monocytes/macrophages. Remarkably, other upregulated overall signaling pathways in the secondary CMML post Azacitidine plus Venecla treatment group are CD40 and CD23, which are considered to be T/NK cell activation molecules. 22–24 The incoming and outgoing signaling patterns analysis in the after-treatment group indicated that similar results compared to overall signaling pathways analysis (Supplementary Figs. 7D and E).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}