Our results provide an interpretation of the already known pro-coagulant pattern of patients with ARDS due to COVID-19 infection, and, to our knowledge, this is the first investigation of the coagulation profile based on markers of thrombin generation and fibrinolysis. Previous studies with viscoelastic tests had already stressed that the main finding in these patients is an abnormally increased clot firmness, with no signs of hyperfibrinolysis or even fibrinolysis shutdown.6, 15-17 However, analyses based on standard or viscoelastic tests remain inconclusive with respect to the nature of this pattern. From this respect, our study suggests an interpretative view of the major factors determining the CoAC, and on their differences in survivors and non-survivors.
Thrombin generation
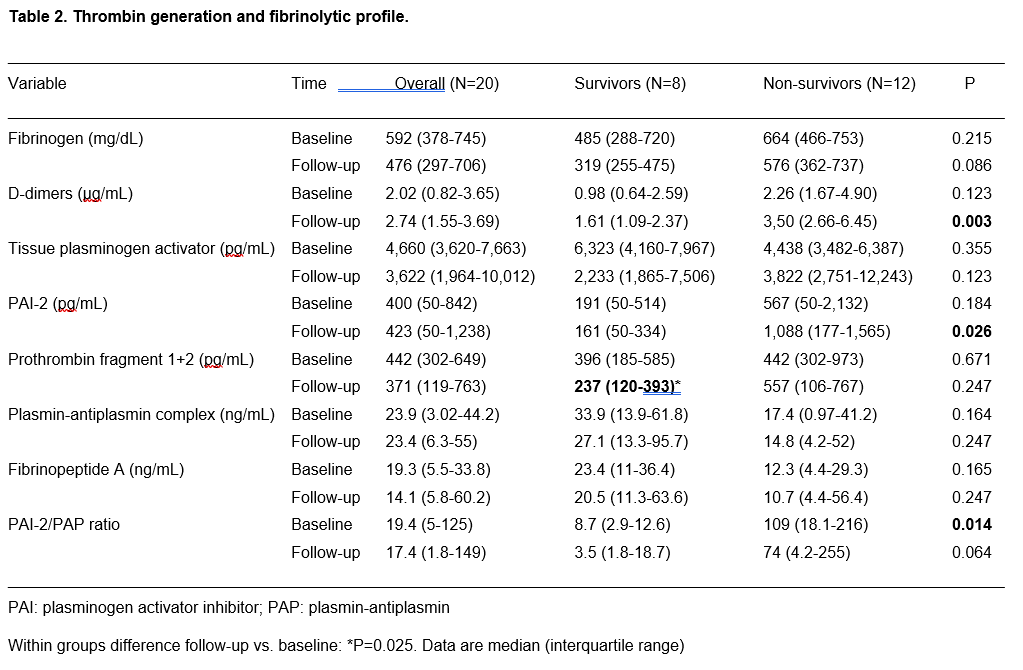
Thrombin generation cannot be assessed with standard or viscoelastic tests. In the first case, variable values of aPTT and PT have been reported1-6, but their changes obviously reflect even the effects of the antithrombotic therapies. In the second, the reaction times (measured with heparinase) did not show a decreased value suggestive for an increased thrombin generation.15,17 We addressed thrombin generation by measuring PF 1+2, a marker of prothrombin cleavage to thrombin. We found values ranging from 20 to over 2,300 pg/mL at baseline (median 442 pg/mL), and from 20 to over 3,300 pg/mL at follow-up (median 371 pg/mL). The normal values of PF 1+2 in healthy subjects is between 11 and 22 pg/mL, and hence a strong thrombin generation is present in COVID-19 ARDS patients. In other models of severe sepsis, median values of 100-200 pg/mL were reported18; in our series, thrombin generation is almost double these values. Of notice, thrombin generation behaved differently in survivors and non-survivors. At baseline, there were no significant differences between groups; however, in survivors, thrombin generation significantly decreased at follow-up, whereas it remained stable or increased in non-survivors.
Fibrinogen and fibrin generation
Elevated fibrinogen levels are confirmed in our patient population, as already highlighted in other studies.6,15,16 Fibrinogen levels are decrease by 35% at follow-up in survivors, and by 16% only in non-survivors. Fibrin generation was addressed by measuring Fibrinopeptide A, a marker of fibrinogen cleavage to fibrin. Normal levels of Fibrinopeptide A range between 0.1 and 2 ng/mL, with a mean at 0.50 ng/mL.19 In our series, Fibrinopeptide A largely exceeded the upper limit of the normal range, both in survivors and non-survivors, at baseline and follow-up, with a trend toward higher values in survivors. Hence, as a logical consequence of the increased thrombin generation, fibrin generation is increased as well, and continues unabated from baseline to follow-up. Fibrinopeptide A increases in patients with bacterial and virus sepsis, as a consequence of the cross-link between inflammation and coagulation. The values found in our series are in the range of what previously found in patterns of bacterial sepsis, severe sepsis, and septic shock.20
Fibrinolysis activation
Tissue plasminogen activator is a fibrinolytic agent released mainly by endothelial cells as a response to fibrin generation. Its normal values are around 10,000 pg/mL21,22, but in conditions of systemic inflammatory reaction syndrome or severe sepsis its values are usually higher (> 10,000 pg/mL)23, with reported values up to 50,000-70,000 pg/mL in non-survivors.24
Quite surprisingly, in our series, the median value of tPA was at the lower limits of normal range both at baseline and follow-up, and both in survivors and non-survivors, with only one case reaching 20,000 pg/mL. Therefore, it apparently seems that fibrinolysis was not activated in these patients, despite an increased thrombin (and fibrin) generation.
Fibrinolysis inhibition
Plasminogen activator inhibitor-2 is a powerful inhibitor of fibrinolysis, released by monocytes. It is usually undetectable in plasma from normal subjects, and it is considered an inhibitor of urokinase-plasminogen activator acting mainly at an extravascular level.24
Due to its ability to act at the level of interstitial tissues (including lung interstitium) and to its non-detectability in normal subjects (except in pregnant women), we have measured PAI-2 as a marker of fibrinolysis inhibition. Previous studies highlighted that in septic patients PAI-2 becomes detectable, with values of 500-1,000 pg/mL in survivors and up to 30,000 pg/mL in non-survivors.24
In our series, elevated values of PAI-2 were observed especially in non-survivors, and at follow-up the level of PAI-2 was 6-folds that of survivors, with a significant between-groups difference. Therefore, fibrinolysis was inhibited, and the extent of inhibition at follow-up was associated with mortality.
The net effect on fibrinolysis
The markers of fibrinolysis in our series were the PAP complexes and the D-Dimers. PAP is a marker of plasmin formation and of plasmin ability to counteract fibrin generation. Not by chance, we could observe a strong relationship between PAP and the marker of fibrin generation Fibropeptide A. Levels of PAP are usually greatly increased under conditions of inflammation and sepsis, with levels exceeding 1,000 ng/mL.25 We could only observe a modest increase of PAP with respect to the reported normal range of 19-27 ng/mL25, more pronounced in survivors than in non-survivors. Therefore, fibrinolysis appears in a shutdown condition, as the result of the balance between increased antifibrinolytic agents (PAI-2) and stable fibrinolytic agents (tPA). This shutdown appears more pronounced in non-survivors, where the PAI-2/PAP ratio is significantly higher than in survivors.
Within this scenario, a particular aspect is represented by D-Dimers behavior. D-Dimers are a fibrin degradation product, and therefore their increase, found both in survivors and (to a larger extent) in non-survivors should be interpreted as marker of increased fibrinolysis. However, contrary to PAP, D-Dimers have no correlation with fibrin generation (as represented by Fibrinopeptide A). Therefore, their raise cannot be ascribed solely to the increased levels of fibrin. Actually, the source of D-Dimers increase in COVID-19 patients is still a matter of debate, and the role of extravascular fibrin degradation (namely in the interstitial and alveolar lung space) has been hypothesized.26 A possible interpretation is that the large concentration of substrate (fibrinogen) generates large amounts of fibrin, and that even in presence of a limited fibrinolysis, the gross amount of fibrin generates high levels of D-Dimers.
The overall picture that can be drawn from our results, is summarized in figure 3.
In both survivors (Panel A) and non-survivors (Panel B) there is an initial phase characterized by the release of proinflammatory cytokines and consequent burst of thrombin generation (reasonably mediated by monocyte and endothelial release of tissue factor). The endothelial cells show a very modest release of tPA. At this stage, both thrombin generation and tPA release do not differ between survivors and non-survivors. Conversely, in non-survivors the release of PAI-2 is higher than in non-survivors, switching the balance between fibrinolysis stimulation and inhibition toward the latter. In both groups there are similar and very high levels of substrate (fibrinogen), and an important increase of fibrin generation. However, fibrinolysis appear more efficient (higher PAP values) in survivors than in non-survivors. In both cases it is likely that an initial thrombi formation may intervene (elevated D-Dimers).
The two pathways clearly diverge at follow-up. In survivors, thrombin generation, PAI-2 and tPA release decrease, as well as fibrinogen levels. Fibrinolysis appears maintained and D-Dimers are stable. Conversely, in non-survivors, thrombin generation and PAI-2 increase, and tPA decreases, with a further shutdown of fibrinolysis and an important increase in D-Dimers. This is likely to represent a condition were thrombi formation may become uncontrolled and clinically relevant.
Thromboembolism, mortality and its predictors, and therapeutic implications
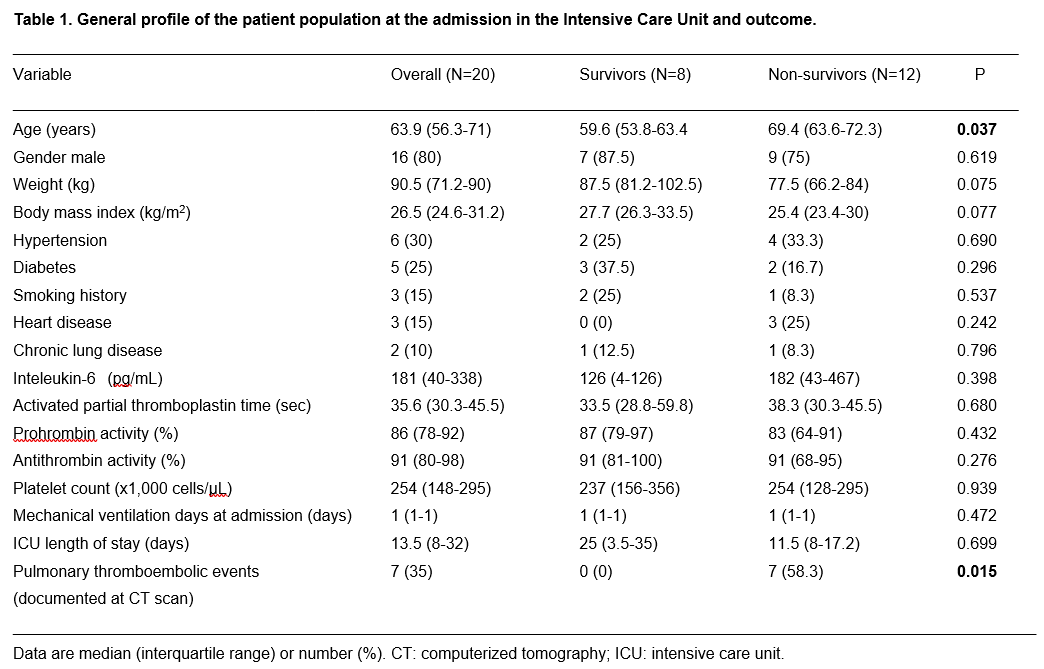
The prothrombotic pattern of CoAC has a relevant clinical impact. Other studies already highlighted the high risk of thromboembolic events in these patients.10, 12-14 It is not the purpose of the present study to address the link between thromboembolic events and mortality. However, it deserves to be quoted that we could observe 7 events of pulmonary thromboembolism (either of minor or major degree) in our patient population, and all of them were diagnosed in patients who lately died. This confirms the uncontrolled thrombi formation in non-survivors, as depicted in figure 3.
The definition of the pathway of CoAC from the onset to the final outcome is of course important providing that (i) an early recognition of patients at high risk of mortality is feasible, and (ii) adequate diagnostic measures and therapies may be established.
With respect to the first issue, our data suggest a high predictive ability of the ratio PAI-2/PAP (cut-off at 12.9) early after the patient is tracheally intubated and placed under mechanical ventilation, and a moderate predictive ability of D-Dimers (cut-off at 1.13 μg/mL). Patients with values above these thresholds deserve a pulmonary CT scan angiography for early detection of micro/macro thrombi.
{kind=link}
{kind=link}