2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin sulfate were sourced from Gibco Life Technologies Co. (Grand Island, NY). The Cell Counting Kit-8 (CCK-8) and ROS Assay Kit - Photo-oxidation Resistant DCFH-DA were obtained from Dojindo Laboratories (Kumamoto, Japan). Glutathione (GSH) Content Assay Kit was obtained from Cayman Chemical Company (United States). Phosphate-buffered saline (PBS) Buffer Premixed Powder (1X) and 10X Tris-Glycine SDS-PAGE Running Buffer were supplied by Sangon Biotech (Shanghai) Co., Ltd (Kudoh et al.). LipoFiter Transfection Reagent Kit were sourced from Hanbio Technology (Shanghai) Co., Ltd. (Shanghai, China). Hoechst, Malondialdehyde (MDA) Content Assay Kit, and 4’,6-Diamidino-2-phenylindole (DAPI) were sourced from Beyotime Institute of Biotechnology (Shanghai, China). HRP Goat Anti-Rabbit IgG (H + L) (AS014), Anti-Nrf2 rabbit (A21508), Anti-NCOA4 rabbit (A5695), Anti-β-actin rabbit mAb (High Dilution) (AC026), HA-Tag rabbit mAb (AE105), DDDDK-Tag rabbit mAb (AE092), Anti-Ferritin Heavy Chain rabbit mAb (A19544), and Anti-LC3B rabbit mAb (A19665) were procured from ABclonal Technology Co. (Shanghai, China). The plasmid was acquired from Genomeditech Co., Ltd. (Kudoh et al.). Prussian Blue Iron Stain Kit was sourced from Beijing Solarbio Science & Technology Co., Ltd (Kudoh et al.). The PrimeScript™ RT-PCR Kit was obtained from Takara Biomedical Technology (Beijing) Co., Ltd. The 2x SYBR Green qPCR Master Mix (Low ROX) was obtained from Selleck.cn (Kudoh et al.).
2.2. Isolation of primary alveolar epithelial cells type Ⅱ
Pregnant C57BL/6J mice at embryonic day 19 (E19) were used for the extraction of primary cells. Under sterile conditions, the uterus was dissected to retrieve the fetal mice. Placing the fetal mice in a laminar flow hood, a horizontal incision was made across the chest of each fetal mouse. The lungs were removed and placed in pre-cooled PBS solution. Care was taken to remove non-lung tissues, such as residual tracheal and connective tissues, as much as possible. The lung tissues were washed three times with PBS, cut into approximately 1 mm3 tissue fragments using ophthalmic scissors, washed once with trypsin solution, and then digested at 37°C for a duration of 20 minutes. To halt the digestion, mix the trypsin with an equal volume of medium with FBS. The mixture was subsequently passed through a 200-mesh sieve. and centrifuged at 1500 revolutions per minute for 5 minutes at low temperature. The liquid above the sediment was removed, and the pellet was retained. The pellet was incubated with 0.1% collagenase I at 37°C for 20 minutes. After digestion, an equal amount of DMEM containing 10% FBS was introduced, with centrifugation for 5 minutes at 1000 rpm. The cell was collected and plated into a culture bottle, which was incubated at 37°C with 5% CO2 for 45 minutes. The liquid above the sediment was then aspirated and transferred to another culture bottle. This same step was repeated for 3 times. Finally, the liquid above the sediment was aspirated, and then centrifuged the cells at 800 rpm for a duration of 5 minutes. After discarding the supernatant, the cell was suspended in a culture medium. and seeded into a culture bottle. After incubating for 24 hours, the medium was exchanged to remove unattached cells, and the culture was continued. The growth and morphology of the cells were examined under an inverted microscope.
2.3. Cell culture
The MLE12 cells, obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), were maintained in a conventional cell culture incubator at 37°C with 5% CO2 and maintained humidity. The cells were maintained in DMEM with 10% FBS and 1% penicillin-streptomycin.
2.4. Oxygen-glucose deprivation and reoxygenation (OGD/R) model
For the OGD/R model induction, cells underwent an initial phase of 8 hours of glucose deprivation, followed by 12 hours of reoxygenation. During the deprivation phase, cells were cultured in a glucose-free medium inside an anaerobic chamber with an atmosphere of 95% N2 and 5% CO2 for 8 hours. Following this, the cells were transitioned to a high-glucose medium and grown in a conventional cell culture incubator for an additional 12 hours to reoxygenate.
2.5. Cell viability assessment
Within a 96-well plate, MLE12 cells were cultured at 1 × 104 cells/well. Once the cells had fully adhered, they were subjected to various treatments. Subsequently, the cells underwent 8 hours of oxygen-glucose deprivation (OGD) followed by 12 hours of reoxygenation to establish the OGD/R model. Upon completion of the model establishment, each well received 10 µL of CCK-8 solution and 90 µL of medium. Following this, the cells were kept for 4 hours in the dark before their optical density (OD) at 450 nm was mrasured by a microplate reader.
2.6. Animals
Twenty-four C57BL/6J mice (6–8 weeks) were acquired from the Animal Center of Shanghai Jiao Tong University School of Medical Sciences. They were kept under regulated environmental conditions, and a 12-hour light/dark cycle, with a temperature of 21 ± 2°C. All experimental procedures were performed following the guidelines stipulated by the National Institutes of Health (NIH) and took permission from the Animal Research Ethics Committee at Shanghai Jiao Tong University (ethical approval number A2023114).
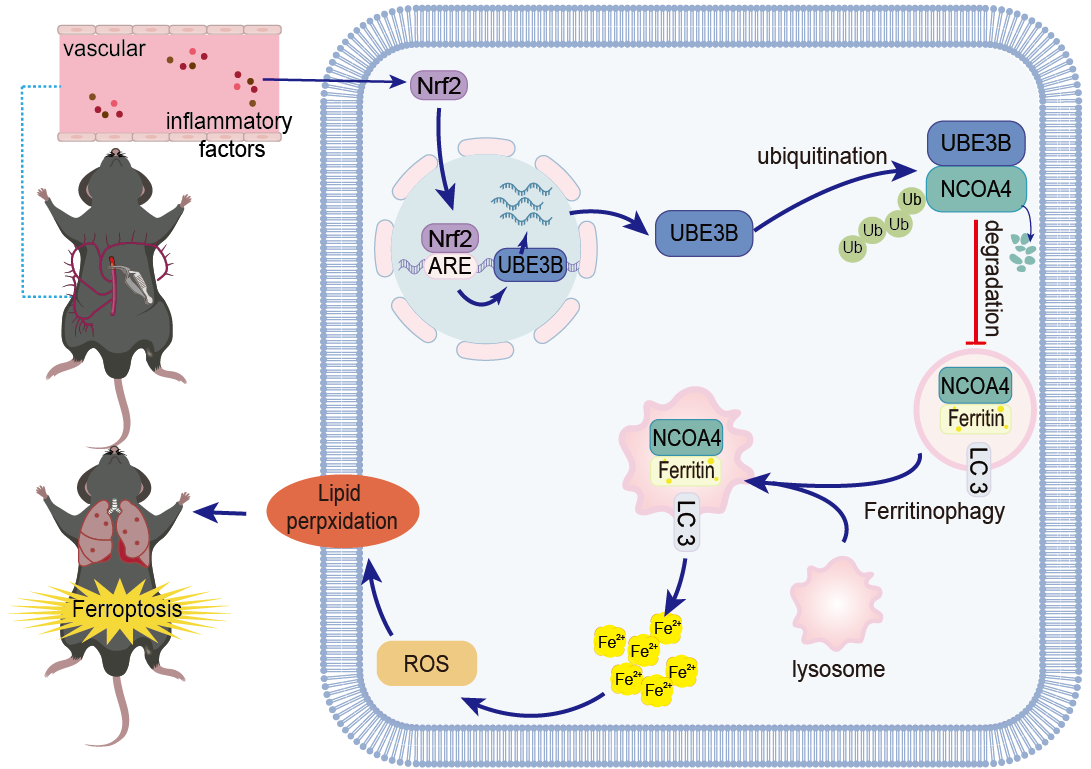
2.7. Intestinal ischemia/reperfusion (II/R) mouse model
Prior to the experiment, animals underwent a 24-hour fasting period with access to water. The mice were anesthetized intraperitoneally with sodium pentobarbital at a dosage of 50 mg/kg and underwent a midline incision. To induce intestinal ischemia, The superior mesenteric artery was temporarily occluded using non-invasive vascular clips for a duration of 45 minutes followed by a 180-minute reperfusion period. Sham control mice underwent identical procedures without vascular occlusion. After 3 hours of reperfusion, the animals were euthanize, and tissue samples were promptly frozen at -80°C.
2.8. Malondialdehyde (MDA) assay(Rui et al., 2021)
Homogenize or lyse the tissues or cells using PBS or lysis buffer. For tissues, the weight of the tissue should constitute 10% of the volume of the homogenization or lysis buffer; for cells, use 0.1 ml of lysis or homogenization buffer per one million cells. After homogenization or lysis, centrifuge at 10,000g to 12,000g for 10 minutes at 4ºC and collect the supernatant. Dilute the appropriate amount of the standard with distilled water to concentrations of 1, 2, 5, 10, 20, and 50µM. In a centrifuge tube or another suitable container, add 0.1 ml of the homogenization buffer, lysis buffer, or PBS as a blank control, 0.1 ml of the standards above different concentrations to create the standard curve, and 0.1 ml of the sample for measurement. Then, add 0.2 ml of the MDA detection working solution. Mix well and heat at 100ºC or in a boiling water bath for 15 minutes. Cool the samples to room temperature in a water bath, then centrifuge at 1000g for 10 minutes. Transfer 200 µl of the supernatant to a 96-well plate, and measure the absorbance at 532 nm using a microplate reader. Calculate the MDA content in the sample solution.
2.9. GSH assay(Wu et al., 2023)
The GSH levels were measured using a GSH assay kit (Cayman Chemical, Catalog No. 703002, Ann Arbor, MI, USA).
2.10. Iron ion assay
Weigh 0.1 g of fresh tissue and mix 0.9 mL of homogenization reagent. Centrifuge the mixture at 10,000 × g for a duration of 10 minutes, and collect the resulting supernatant. Dilute the sample to a concentration within the linear range of the kit (0.4–50 µmol/L). Prepare standard tubes by transferring 300 µL of different concentrations of the standard solution into corresponding 1.5 mL EP tubes. Similarly, prepare test tubes by transferring 300 µL of the sample into corresponding 1.5 mL tubes. Mix 150 µL of reagent two to each tube, mix thoroughly, and place the mixture in an incubator set at 37°C for a duration of 10 minutes. Centrifuge all tubes at 12,000 × g for 10 minutes. Transfer 300 µL of the supernatant from each tube into the respective wells of a 96-well plate. Their OD were measured at 593 nm in each well by a microplate reader.
2.11. Perls staining
After preheating the paraffin sections, deparaffinize twice in xylene for 5 minutes each time. Gradually rehydrate with ethanol, 3 minutes per grade, and rinse with distilled water for 3 minutes.
Thaw the frozen sections, allow them to equilibrate at 4°C for 10 minutes, and then rinse them with distilled water for 3 minutes. For cell smears, fix with pre-cooled low-grade alcohol for 3 minutes or 4% tissue cell fixative for 20 minutes, followed by two washes with 70% ethanol for 1 minute each, and two rinses with distilled water for 2 minutes each. Prepare Perls staining working solution before use, add it to cover the sections, stain for a duration of 30 minutes, and rinse with distilled water for 10 minutes. For nuclear fast red staining, stain for 5–10 minutes, rinse quickly with tap water for 2–3 seconds. Dip in 75%, 85%, 95% ethanol for 3–5 seconds each, and twice in 100% ethanol, each time for 1 minute. Clear with xylene or environmentally friendly tissue deparaffinization solution twice, each time for 1–2 minutes. Seal with neutral gum.
2.12. Lung injury score
The mice lung tissues underwent fixation in 10% formalin, and embedding in paraffin and sectioning into 5 µm slices. Thereafter, the slices were stained with hematoxylin and eosin (HE) and examined using a Nikon-Ni-U light microscope equipped with an Optronics DEI-470 digital camera. To evaluate acute lung injury (ALI), a five-point scoring system (0–4) was utilized based on several parameters: The scoring criteria included assessment of alveolar and interstitial edema, intra-alveolar inflammatory cell infiltration, alveolar hemorrhage, and atelectasis, which were graded as follows:
Grade 0: Normal morphology, with < 15% tissue occupancy and > 85% alveolar space occupancy.
Grade 1: Mild injury, with 15–25% tissue occupancy and 75–85% alveolar space occupancy.
Grade 2: Moderate injury, with 25–50% tissue occupancy and 50–75% alveolar space occupancy.
Grade 3: Severe injury, with 50–75% tissue occupancy and 25–50% alveolar space occupancy.
Grade 4: Very severe injury, with 75–100% tissue occupancy and 0–25% alveolar space occupancy.
2.13. Co-immunoprecipitation (CO-IP) assay
Tissue sample preparation: Rinse the tissue with PBS, add pre-chilled RIPA buffer (mild RIPA, 1000 µL/100 mg tissue, supplemented with 10 µL protease inhibitor/1000 µL RIPA), and keep on ice for 10 minutes. Take two sterilized steel beads and grind the tissue. Centrifuge at 4°C, 12000 rpm for a duration of 15 minutes, and retrieve the liquid. Determine protein concentration using the BCA method. For total protein, take 500 ul and add 10 µL of Nrf2 antibody; take 300 µL and add IgG antibody. Incubate overnight at 4°C with slow inverting. The remaining protein is combined with loading buffer and heated to 100°C, boiling for 15 minutes. Pre-wash Protein A beads: Resuspend Protein A + G magnetic beads by gently pipetting, transfer an appropriate amount into a clean centrifuge tube and adjust to a final volume of approximately 0.5 ml with 1X TBST. Gently resuspend Protein A + G magnetic beads by pipetting. Put the tube on a magnetic rack for 10 seconds to allow the beads to adhere to the magnet, then gently discard the supernatant. Perform this washing step two additional times. Next, perform magnetic separation using the Protein A + G magnetic beads: discard the supernatant, add the sample, resuspend the beads, and allow the mixture to incubate for 30 minutes on a rocking mixer. Add 500 µL of 1X TBST, gently resuspend Protein A + G magnetic beads by pipetting, place on a magnetic stand for 10 seconds, discard the liquid above, and repeat washing three times. For every 10–20 µL of the original magnetic bead volume, mix 10 µL 2X SDS-PAGE loading buffer. Heat the mixture at 100°C for 5 minutes, then place it on a magnetic stand for 10 seconds to separate the beads and collect the supernatant. Analyze by Western Blotting to confirm the binding proteins.
2.14. Western blot
Proteins were blended with SDS-PAGE loading buffer and heated at 95°C for about 15 minutes. The proteins underwent electrophoresis on a 10% SDS-PAGE gel and transferred onto a polyvinylidene fluoride (PVDF) membrane. In succession, the membrane was immersed in TBST solution with 10% skimmed milk for 1.5 hours, subsequent to 3 washes with TBST. Primary antibodies against Nrf2 (Rabbit, 1:500), NCOA4 (Rabbit, 1:1000), UBE3B (Rabbit, 1:1000), FTH1 (Rabbit, 1:1000), LC3 (Rabbit, 1:1000), and β-actin (Rabbit, 1:100000) were then incubated with the membrane overnight at 4°C. On the subsequent day, the protein bands were rinsed 3 times with TBST before being exposed to secondary antibodies (HRP-goat anti-rabbit, 1:5000) for 1 hour. Hereinafter 3 washes with TBST, bands were detected with an enhanced ECL chemiluminescent substrate kit and quantified by the Bio-Rad multicolor fluorescence and chemiluminescence imager.
2.15. Immunofluorescence assay
Cryosections of the lungs were prepared and treated with 0.3% Triton-100 for 15 minutes to permeabilize them, followed by a 2-hour incubation with 3% BSA. And the sections were exposed separately to primary antibodies targeting NCOA4, UBE3B, FTH1, and LC3, and incubated overnight at 4°C. With overnight exposure, the sections were rinsed with five washes of PBS. To visualize the target proteins, the sections underwent incubation with fluorescently labeled secondary antibodies for 1 hour. Subsequently, three washes with PBS were performed. Finally, the sections were counterstained with DAPI to observe cell nuclei and captured by a confocal laser scanning microscope (Carl Zeiss LSM710, Germany).
2.16. Plasmid transfection
To initiate plasmid transfection, cells were seeded in a 6-well plate the previous day at about 1 × 106 cells/well. The cells grew until they reached about 70% confluence the following day. Before the transfection procedure described below, the culture medium was changed to 2 mL of fresh culture medium containing serum. In a sterile and clean centrifuge tube, 125 µL of serum-free DMEM culture medium (devoid of antibiotics) was added for each well slated for transfection. Subsequently, 2 µg of plasmid DNA was introduced into each tube, and gentle pipetting facilitated thorough mixing. Following this, 4 µL of LipoFiter transfection reagent was added, and the mixture was gently swirled to ensure homogeneity. Whether dealing with adherent or suspended cells, the 125 µL Lipo8000™ transfection reagent-DNA concoction was evenly dispensed into each well of the six-well plate, with careful swirling to guarantee uniform distribution. The cells were then allowed to incubate for approximately 72 hours before sample collection.
2.17. Quantitative real-time PCR assay
Cells or tissues were treated with TRIzol to extract RNA. The isolated RNA was subsequently converted in reverse to generate cDNA by using the PrimeScript™ RT-PCR Kit. Subsequent qPCR was conducted with 2x SYBR Green qPCR Master Mix. The primer sequences for each target are as follows:
Mouse_actin, beta (β-actin)
Forward primer: GCTCACCATTCACCATCTTGTCTTG
Reverse primer: TCCTGCTTGCTGATCCACATCTG
Mouse_Nrf2
Forward primer: TTGCCACCGCCAGGACTACA
Reverse primer: AACTTGTACCGCCTCGTCTGGA
Mouse_nuclear receptor coactivator 4 (Ncoa4)
Forward Primer: GCCAGAGCAGAAGTCAGCATCC
Reverse Primer: GCCAGTCCTGTGGGTTGGTACT
Mouse_ferritin heavy polypeptide 1 (FTH1)
Forward primer: CCATCAACCGCCAGATCAACCT
Reverse primer: GCAAAGTTCTTCAGAGCCACATCAT
Mouse_microtubule-associated protein 1 light chain 3 beta
Forward primer: CAAGCCTTCTTCCTCCTGGTGA
Reverse primer: TTGCTGTCCCGAATGTCTCCTG
2.18. Transmission electron microscopy (TEM)
The mice lung tissues were preserved in 0.05 M sodium cacodylate buffer with 2.5% glutaraldehyde at pH 7.2 at 25°C for a duration of 2 hours. Following this, they were dealt with 2% OsO4 in 0.1 M sodium cacodylate buffer for an additional 2 hours, followed by immersion in 1% aqueous uranyl acetate for a period of 18 hours. With the dehydration in ethanol, the samples were preserved in resin, sectioned using copper grids, and stained with lead citrate and uranyl acetate. Visualization was carried out by a transmission electron microscope (FEI Company, Oregon).
2.19. Statistical Analysis
The mean values with their standard deviations (SD) are presented. Statistical analyses were carried out by using Prism software (version 9.0.0), employing one-way analysis of variance (ANOVA) for between-group comparisons. Statistical significance was shown as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
{kind=link}