Bacterial strains and host matrix
Defibrinated sheep blood (E&O Laboratories, UK) and EDTA-treated whole blood from healthy human volunteers were used as model host environment matrix. The list of clinical and/or American Type Culture Collection (ATCC) strains tested/used in this study can be found in Table 1, S1, S2 and S3.
Primers, probes, and amplification parameters
To assess the depletion efficiency of the protocols, universal 16S rRNA, 18S rRNA and bacterial species-specific primers and probes were utilised [9–12]. The total volume of individual qPCR reactions was 20 µL, with 2x Rotor-Gene Multiplex PCR master mix (Qiagen, Hilden, Germany) and 2 µL DNA template. qPCR reactions were performed on a Rotor-Gene Q real-time cycler for up to 40 cycles (Qiagen, Hilden, Germany). The primer/probe sequences, concentrations and PCR cycling parameters are provided in Tab. S4.
Benchmarking host depletion kits and protocols
A chemical host DNA depletion protocol, named M-15, was developed in this study (Fig. 1, Supplement Protocol: M-15 mNGS) and benchmarked alongside two commercial [Zymo-HostZERO (named C1; Zymo Research, CA, USA), MolYsis-Complete5 (C2; Molzym GmbH & Co, Bremen, Germany)] and three published host depletion protocols P1 [6], P2 [8], and P3 [7].
To determine host depletion efficiency and analytical sensitivity of the workflows, serially diluted (100 to 105 CFU/mL) exponential growth phase cells of Escherichia coli (ATCC 25922), Staphylococcus aureus (ATCC 25923), and Pseudomonas aeruginosa (ATCC 27853) strains were spiked in 400 µL sterile whole blood (human and sheep) and processed to deplete host cells/DNA with all the six protocols. Following host depletion with P1, P2, P3 and M-15, DNA extraction was performed using QIAamp UCP Pathogen Mini Kit (Qiagen, Hilden, Germany) with the recommended homogenisation step by bead beating (Pathogen Lysis Tubes L, Qiagen, Hilden, Germany). For protocols C1 and C2, DNA extraction was performed using the reagents supplied with the kits. Total DNA was extracted from the same spiked blood samples without host depletion similarly as unenriched (non-CHDD) controls. Extracted DNA was tested using qPCR with the 16S rRNA, 18S rRNA, and species-specific primers/probes described above.
In addition, a subset of the simulated M-15 CHDD samples, containing 104 to 101 CFU/mL blood was extracted, amplified and barcoded for sequencing using ONT Rapid PCR barcoding kit (SQK-RPB004; Oxford Nanopore technologies, Oxford, UK) and sequenced on MinION R9.4.1 flow cells. Live GPU basecalling was performed in high accuracy mode (dna_r9.4.1_450bps_hac) and passed Fastq reads were analysed using Chan Zuckerberg infectious diseases (CZ ID) (v0.7; https://czid.org/) Nanopore workflow (last accessed: 07/08/2024) [13].
Determining the contribution of host depletion on bacterial cell loss/viability
Bacterial cell loss and/or viability following host depletion was determined using E. coli, S. aureus, and P. aeruginosa ATCC strains. Approximately 30 and 3 CFU of the exponential phase cells were spiked in 400 µL sterile whole blood, and the samples were processed to deplete host DNA with three top performing protocols C1, C2, P1, and M-15. Host depleted samples were resuspended in PBS and plated on nutrient and blood agar media to estimate bacterial concentrations using the method described by Miles & Misra [14]. The same samples were plated directly without host depletion as controls. All the inoculated plates were incubated overnight at 37°C, and the number of visible colonies was recorded the next day.
Rapid M-15 mNGS workflow tested on culture enriched specimens
To determine the efficiency of M-15 mNGS protocol on culture enriched samples, six culture enriched suspected BSI samples were tested initially. Three samples flagged positive on BACT/ALERT (bioMérieux, Marcy-l'Étoile, France) and the remaining three were reported to be culture negative. The same samples were processed directly without M-15 as no depletion controls. To minimise hands on time, rapid alkaline cell lysis was performed using 4 µL direct or host depleted samples resuspended in 1 mL PBS using REPLI-g single cell kit (Qiagen, Hilden, Germany). Multiple displacement amplification (MDA) was performed with the same kit for 60 minutes according to the manufacturer’s recommendation. Amplified MDA products were then debranched for 10 minutes at 37° C using T7 endonuclease (New England Biolabs, MA, United States) and subjected to 0.5x AMPure bead cleanup. Library was prepared using ONT Rapid barcoding kit (SQK-RBK004) with 400 ng amplified debranched DNA product/sample as an input (Supplement Protocol: M-15 mNGS + low-medium yield protocol). The overall sample processing time of this pilot version of the workflow is approximately 4 hours (processing 6–10 samples), including 80 minutes for host depletion, 20 minutes for DNA extraction (alkaline lysis method), 60 minutes of WGA using REPLI-g Single Cell Kit, and 90 minutes for debranching, cleanup, library preparation and flow cell loading.
To assess the efficiency of adaptive sampling (AS) with or without CHDD with M-15, rapid adapter ligated pooled libraries (six CHDD and six no CHDD) were loaded on a MinION R9.4.1 flow cell and the total number of channels (N = 512) available for sequencing were divided into two parts from the advanced option menu on the MinKNOW graphical user interface. Channels 1 to 256 were chosen for human DNA depletion using T2T-CHM13v2.0 as reference [15] and channels 257–512 for no-adaptive sampling control.
High yield M-15 mNGS workflow validated with culture-positive suspected BSI specimens
An additional 27 blood culture positive samples were tested using the optimised workflow for improved sequencing yield. Chemical host depletion, DNA extraction, MDA (60 minutes) and debranching were performed similarly to the pilot protocol. Only this time, library preparation was performed using Ligation Sequencing Kit (SQK-LSK109) with native barcodes (EXP-NBD104) with 400 ng amplified debranched DNA product/sample as an input (Supplement Protocol: M-15 mNGS + high yield protocol). The overall sample processing time of this optimised workflow takes less than 6 hours (with 6–10 samples) including 80 minutes of CHDD, 20 minutes for DNA extraction (alkaline lysis method), 60 minutes of WGA using REPLI-g Single Cell Kit, and about 180 minutes for debranching, library preparation and loading.
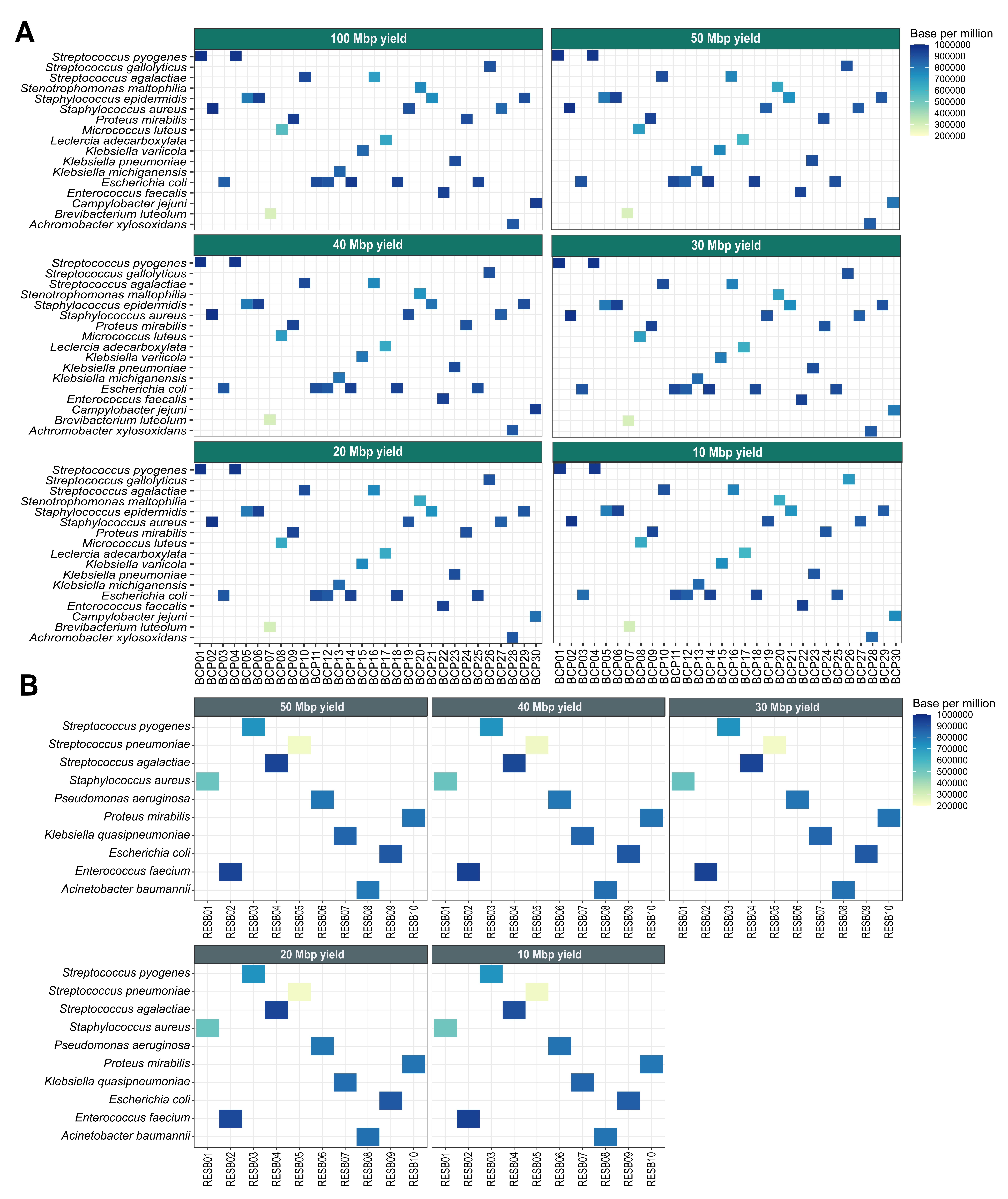
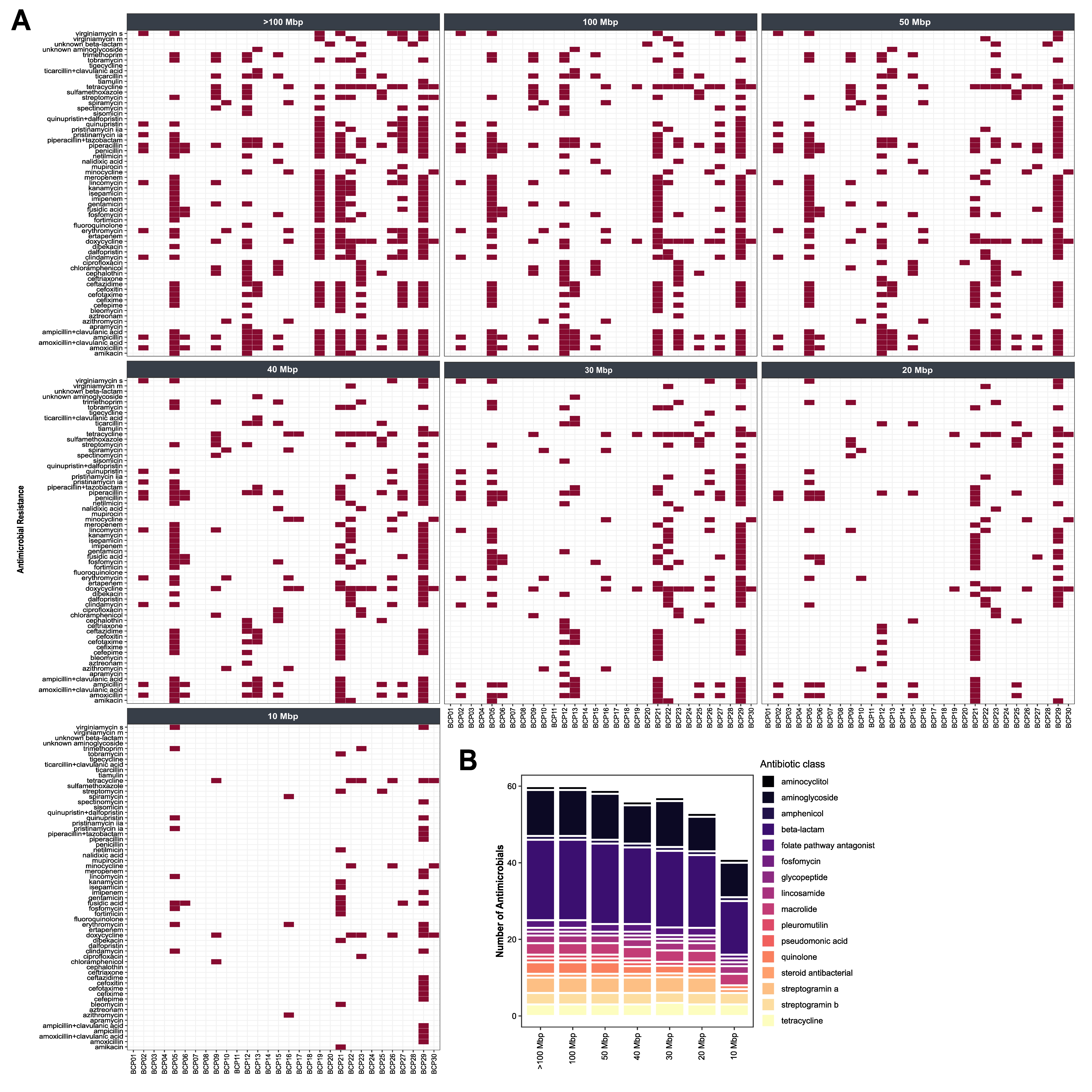
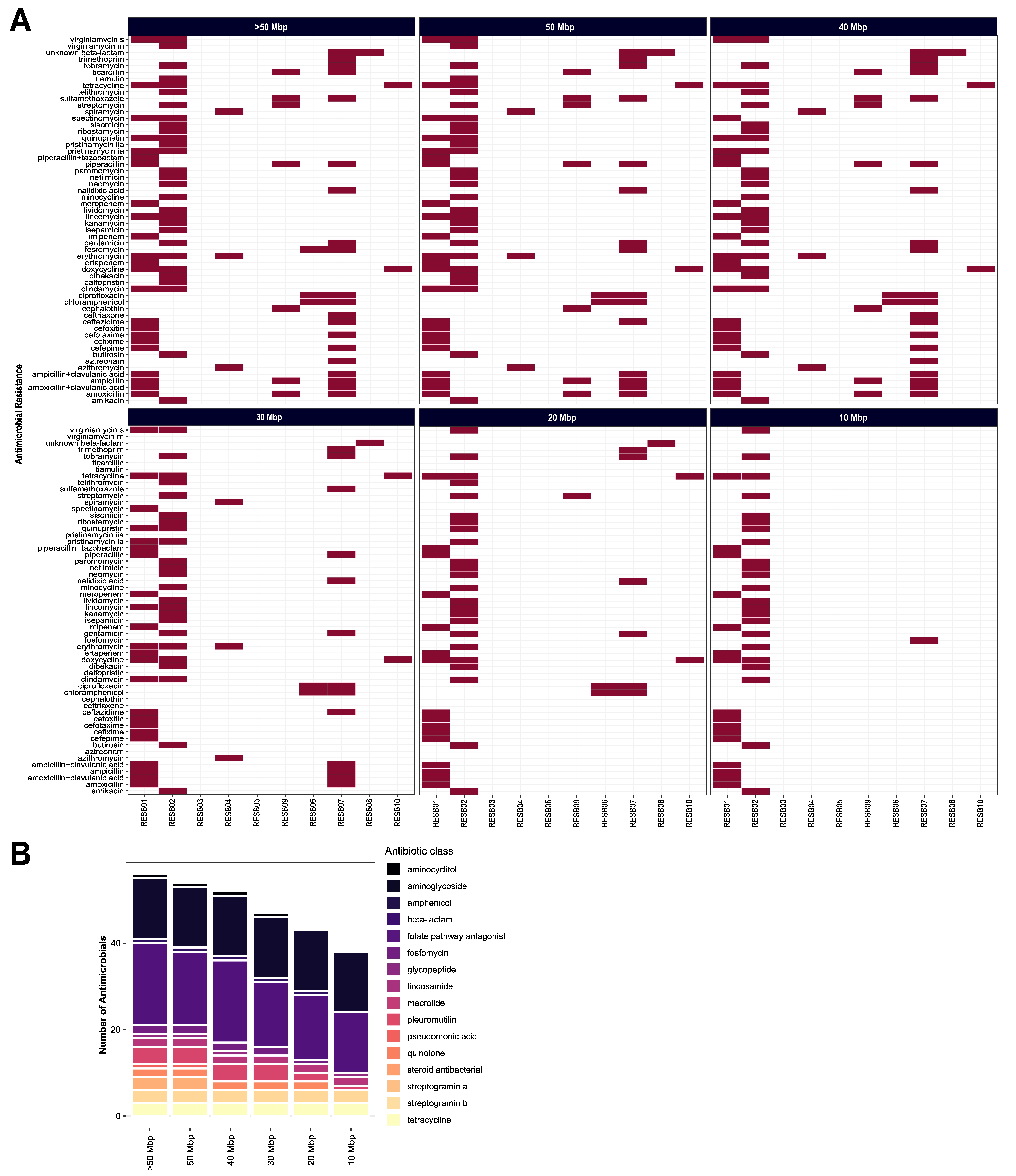
To identify bacterial species, CZ ID pipeline was utilised as mentioned above with a 20% abundance cutoff (200,000 Base Per Million; BPM for CZ ID). Host-filtered contigs remaining after the 20% abundance cutoff were analysed to identify AMR determinants with ResFinder 4.5.0 [16] using the default options. In addition, host filtered fastq reads remaining after 20% abundance cutoff were mapped against bacterial reference sequences using minimap2[17] to estimate the breadth of coverage and sequencing depth. To determine the minimum sequencing yield required to identify bacterial species and AMR determinants, 100 Mbp, 50 Mbp, 40 Mbp, 30 Mbp, 20 Mbp, and 10 Mbp raw fastq reads were randomly subsampled using rasusa [18] and processed similarly with CZ ID and ResFinder.
Rapid culture enrichment to accelerate microbiological diagnosis
A shorter culture enrichment method was tested with 16 ATCC/clinical isolates representing eight Gram negative and seven Gram positive bacterial species. Approximately 1–10 CFU of actively growing (log phase) bacterial cells were spiked in BD BACTEC™ Standard Blood Culture media (Becton, Dickinson and Company, NJ, USA) supplemented with 10 mL sterile whole blood (sheep), and then subjected to incubation. Culture bottles were taken out at 2-hour intervals over a span of 10 hours and subsequently plated on nutrient and blood agar media using the Miles & Misra method.[14] All the plates were incubated at 37°C overnight and the number of visible colonies was recorded the next day. One mL culture-enriched samples were preserved at -80°C during each sampling session to be tested later. Finally, a subset (10/16) of the 8-hour culture-enriched samples (5 Gram-positive and 5 Gram-negative species) were chosen and processed with the high yield M-15 mNGS protocol (Supplement Protocol: M-15 mNGS + high yield protocol). Species, host: pathogen ratio, antimicrobial resistance genes and minimum DNA yield per sample for identification were determined using the same pipelines and cutoffs mentioned above.
Conventional tests for detecting species, AMR, and predicting AMR with sequencing
In the routine diagnostic microbiology laboratory, 5–10 mL EDTA treated blood samples collected from suspected individuals were incubated in the BACT/ALERT system for up to 5 days. Samples that flagged positive were taken out and inoculated onto appropriate agars, and organisms grown identified using MALDI-TOF mass spectrometry (Bruker Corporation, MA, USA). Phenotypic antimicrobial sensitivity was determined using disc diffusion as described by European Committee on Antimicrobial Susceptibility Testing (EUCAST) guideline [19] or automated systems, e.g. Vitek2 (bioMérieux, Marcy-l'Étoile, France).
To compare routine phenotypic sensitivity and genotypic antimicrobial susceptibility results, AST results for the BACT/ALERT positive BSI samples were compared to ResFinder 4.5.0 phenotype table results. Because ResFinder provides AMR predictions for 92 antimicrobial agents from 21 antibiotic classes, but only predicts resistance or susceptibility (not Intermediate results), only the phenotypic results common between the two (culture and sequencing) and classified as resistant or susceptible were compared. For rapid culture enriched spiked blood samples, for which AST results were not available, WGS (where available) and M-15 mNGS data of the ten ATCC/clinical isolates were compared similarly using ResFinder 4.5.0 phenotype table. Categorical agreement, major error and very major error were calculated using the following formula [20]:
$$\:\text{C}\text{a}\text{t}\text{e}\text{g}\text{o}\text{r}\text{i}\text{c}\text{a}\text{l}\:\text{a}\text{g}\text{r}\text{e}\text{e}\text{m}\text{e}\text{n}\text{t}=\frac{\text{T}\text{R}+\:\text{T}\text{S}}{\text{T}\text{R}+\:\text{T}\text{S}+\text{F}\text{R}+\text{F}\text{S}\:}$$
$$\:\text{M}\text{a}\text{j}\text{o}\text{r}\:\text{e}\text{r}\text{r}\text{o}\text{r}=\frac{\text{F}\text{R}}{\text{F}\text{R}+\text{T}\text{S}}$$
$$\:\text{V}\text{e}\text{r}\text{y}\:\text{m}\text{a}\text{j}\text{o}\text{r}\:\text{e}\text{r}\text{r}\text{o}\text{r}=\frac{\text{F}\text{S}}{\text{F}\text{S}+\text{T}\text{R}}$$
Where: TR = True resistant, TS = True susceptible, FR = False resistant, FS = False susceptible, and where phenotypic AST is taken as the gold standard.
Statistical analysis
R studio 2023.12.1 with R base version 4.3.3 were used in combination with readxl, dplyr, reshape2, ggplot2, and gridExtra packages for performing data cleanup, formatting, analysis and generating graphs.
{kind=link}
{kind=link}
{kind=link}