Methodological overview and design rationale
We designed GOcats with a biologist user in mind, who may not be aware of the dangers associated with using different versions of GO for organizing terms with tools like M2S or how to circumvent potential pitfalls. For instance, although the M2S documentation (https://github.com/owlcollab/owltools/wiki/Map2Slim) states, "We recommend the go-basic version of the ontology be used, which contains: subClassOf (is a), part of, regulates (+ positively and negatively regulates)" and, "You can also use the full version of GO and filter those relationships you do not want to consider," a non-bioinformatician may not be aware of how to filter out relationships from GO in a way that is safe to use the tool—or, more pertinently—the user may wish to use a fuller extent of the information contained in the ontology when organizing their terms. Currently, GOcats version 1.1.4 can handle go-core’s is_a, part_of, and has_part relations, with the has_part reinterpreted to retain proper scoping semantics, as detailed below and elsewhere (26). As the development of GOcats progresses, we plan on handling the organization of terms connected by additional relations such as negatively/positively_regulates.
GOcats uses the go-core version of the GO database, which contains relations that connect the separate ontologies and may point away from the root of the ontology. GOcats can either exclude non-scoping relations or invert has_part directionality into a part_of_some interpretation, maintaining the acyclicity of the graph. Therefore, it can represent go-core as a DAG.
GOcats is a Python package written in major version 3 of the Python program language (25) and available on GitHub and the Python Package Index. It uses a Visitor design pattern implementation (27) to parse the go-core Ontology database file (4). Searching with user-specified sets of keywords for each category, GOcats extracts subgraphs of the GO DAG and identifies a representative node for each category in question and whose child nodes are detailed features of the components. Figure 7 illustrates this approach, and details follow in pseudocode.
To overcome issues regarding scoping ambiguity among mereological relations, we assigned properties indicating which term was broader in scope and which term was narrower in scope to each edge object created from each of the scope-relevant relations in GO. For example, in the node pair connected by a part_of or is_a edge, node 1 is narrower in scope than node 2. Conversely, node 1 is broader in scope than node 2 when connected by a has_part edge. This edge is therefore reinterpreted by GOcats as part_of_some. This reinterpretation is not meant to imply exclusivity in composition between the meronym and the holonym. It simply stands as a distinction between “part of all” which is what the current “part_of” relationship implies, and “part of some,” or to be more verbose “instance a is part of instance b in at least one known biological example.” We have described additional explanations and rationale for this re-interpretation elsewhere and demonstrate improvement in annotation enrichment statistics when this re-interpretation is used (26).
While the default scoping relations in GOcats are is_a, part_of, and has_part, the user has the option to define the scoping relation set. For instance, one can create go-basic-like subgraphs from a go-core version ontology by limiting to only those relations contained in go-basic. For convenience, we have added a command line option, “go-basic-scoping,” which allows only nodes with is_a and part_of relations to be extracted from the graph. Detailed API documentation and user-friendly tutorials are available online (https://gocats.readthedocs.io/en/latest/).
For mapping purposes, Python dictionaries are created which map GO terms to their corresponding category or categories. For inter-subgraph analysis, another Python dictionary is created which maps each category to a list of all its graph members. By default, fine-grained terms do not map to category root-nodes that define a subgraph that is a superset of a category with a root-node nearer to the term. For example, a member of the “nucleolus” subgraph would map only to “nucleolus,” and not to both “nucleolus” and “nucleus”. However, the user also has the option to override this functionality if desired with a simple “--map-supersets” command line option. Furthermore, we’ve included the option for users to directly input GO terms as category representatives, should they not wish to use keywords to define subgraph categories. This is helpful for users who have already compiled lists of GO terms by hand for use with other tools.
Implementation overview
As illustrated in the UML diagram in Figure 8A, the GOcats package is implemented using several modules that have clear dependencies starting from a command line interface (CLI) in gocats.py which depend on most of the other modules including ontologyparser.py, godag.py, subdag.py and tools.py. GOcats uses 10 classes implemented across ontologyparser.py, godag.py, subdag.py, and dag.py to extract and internally represent the GO database. GoParser, which inherits from the base OboParser class (Figure 8B), utilizes a visitor design pattern and regular expressions to parse the flat GO database obo file and instantiate the objects necessary to represent the GO DAG structure. These instantiated objects include (Figure 8C): 1) the GoGraph container object for the parts of the graph, which inherits from a more generic OboGraph containing functions for adding, removing, and modifying nodes and edges; 2) GoGraphNode objects for representing each term parsed from the ontology, which inherits from AbstractNode; 3) AbstractEdge objects for representing each instance of a relation parsed from the ontology; and 4) DirectionalRelationship objects, which inherit from the more generic AbstractRelationship object for representing each type of directional relation encountered in the ontology (for GO, all relations are directional, and this distinction is made only in anticipation for future extensions to handle other ontologies).
AbstractEdge objects and AbstractNode objects contain references to one another, which simplifies the process of iterating through ancestor and descendant nodes and allows for functions such as AbstractEdge.connect_nodes, which requires that the edge object update the node object’s child_node_set and parent_node_set. In this context, AbstractNode is a true abstract base class, while AbstractEdge started out as an abstract base class but eventually became a concrete class during development. However, we see the possibility of AbstractEdge becoming a base class in the future.
Ancestors and descendants of a node are implemented as sets, which are lazily created through the use of a Python property decorator (i.e. Python’s preferred “getter” syntax). At the first access of these sets through the ancestor or descendent property, the set is calculated with a recursive algorithm, stored for future use, and returned for immediate access. Subsequent accesses simply return the stored set. If the set of edges within a node change, the ancestor and descendent node sets will be recalculated on their next access. This implementation prevents pre-calculation of these sets when they are not used, while enabling their reuse within efficient graph analysis methods.
AbstractEdge also contains a reference to a DirectionalRelationship object, which is critical for graph traversal. This is because DirectionalRelationship contains the true directionality of the mereological correspondence between the categorization relevant relations (is_a, part_of, and has_part). In other words, it is within this class that we define in which direction the edge should be traversed when categorizing terms. Currently these rules are hard-coded within GoParser’s relationship_mapping dictionary.
The gocats.py module (Figure 8A) implements the command line interface and is responsible for handling the command line arguments, using the provided keywords and specified arguments like namespace filters (e.g. Cellular Component, Molecular Function, and Biological Process) to instantiate a GoParser object, a GoGraph object and a SubGraph object for each set of provided keywords. After creation of the GoGraph internal representation, each category subgraph is created by first instantiating the SubGraph object and calling the from_filtered_graph function, which filters to those nodes from the GoGraph containing the keywords in their names and definition. Note that the SubGraph object and GoGraph object both inherit from OboGraph, and that the SubGraph object contains a reference to GoGraph object (supergraph data member) of which it is a subgraph. This design was implemented to avoid accidental alterations of the GoGraph object when altering the contents of the subgraph, and to allow for specialization of functions within SubGraph without needing to use unique names e.g. add_node(). GoGraphNode objects within the subgraph are wrapped by SubGraphNode objects, which are directly used by the SubGraph object, but retain all original properties such as name, definition, and sets of edge object references, otherwise insidious changes could occur to the GoGraph object when updating the SubGraph object. The SubGraph object also contains a CategoryNode object, which wraps the category representative GoGraphNode object(s) for the subgraph category.
Specific implementation details
User-provided keyword sets are used by GOcats to query GO terms’ name and definition fields to create an initial seeding of the subgraph with terms that contain at least one keyword. This seeding is a list of nodes from the whole go-core graph (supergraph) that pass the query. Node synonyms were not used, due to there being four types of synonyms in GO: exact, narrow, broad, and related. Also, many nodes within GO do not have synonyms, which may create an unequal utilization of nodes if synonyms were queried. However, in the future, synonym utilization for seeding purposes may be revisited.
FOR node in supergraph.nodes
IF keyword from keyword_list in node.name or node.definition
APPEND node to subgraph.seeding_list
Using the graph structure of GO, edges between these seed nodes are faithfully recreated except where edges link to a node that does not exist in the set of newly seeded GO terms. During this process, edges of appropriate scoping relations are used to create children and parent node sets for each node.
FOR edge in supergraph.edges
IF edge.parent_node in subgraph.nodes AND /
edge.child_node in subgraph.nodes AND /
edge.relation is TYPE: SCOPING
APPEND edge to subgraph.edges
ELSE
PASS
FOR subnode in subgraph.nodes
subnode.child_node_set = {child_node for child_node in /
supergraph.id_index[subnode.id].child_node_set if /
child_node.id in subgraph.id_index}
subnode.parent_node_set = {parent_node for parent_node in /
supergraph.id_index[subnode.id].parent_node_set if /
parent_node.id in subgraph.id_index}
GOcats then selects a category representative node to represent the subgraph. To do this, a list of candidate representative nodes is compiled from non-leaf nodes, i.e. root-nodes in the subgraph which have at least one keyword in the term name. A single category representative root-node is selected by recursively counting the number of children each candidate term has and choosing the term with the most children.
FOR subnode in subgraph
IF subnode.child_node_set != None AND ANY keyword in /
subnode.name
candidate_list.append(subnode)
ELSE
PASS
representative_node = MAX(LEN(node.descendants) FOR node in /
candidates)
Because it may be possible that highly-specific or uncommon features included in the GO may not contain a keyword in its name or definition but still may be part of the subgraph in question by the GO graph structure, GOcats re-traces the supergraph to find various node paths that reach the representative node. We have implemented two methods for this subgraph extension: i) comprehensive extension, whereby all supergraph descendants of the representative node are added to the subgraph and ii) conservative extension, whereby the supergraph is checked for intermediate nodes between subgraph leaf nodes and the subgraph representative node that may not have seeded in the initial step.
Comprehensive extension:
FOR node in supergraph
IF ANY (ancestor_node in node.ancestors) in subgraph
subgraph_nodes.append(ancestor_node)
UPDATE subgraph # appropriate edges added and parent/child nodes
# assigned
Conservative extension:
FOR leaf_node in subgraph.leaf_nodes # nodes with no children
start_node = leaf_node
end_node = representative_node
FOR node in super_graph.start_node.ancestors ∩ /
supergraph.end_node.descendents
subgraph_nodes.append(node)
UPDATE subgraph # appropriate edges added and parent/child nodes
# assigned
The subgraph is finally constrained to the descendants of the representative node in the subgraph. This excludes unrelated terms that were seeded by the keyword search due to serendipitous keyword matching.
Creating category mappings from UniProt’s subcellular location controlled vocabulary
We created mappings from fine-grained to general locations in UniProt’s subcellular location CV (2) for comparison to GOcats. To accomplish this, we parsed and recreated the graph structure of UniProt’s subcellular locations CV file (13) in a manner similar to the parsing of GO (Figure 2). Briefly, the flat-file representation of the CV file is parsed line-by-line and each term is stored in a dictionary along with information about its graph neighbors as well as its cross-referenced GO identifier. We assumed that terms without parent nodes in this graph are category-defining root-nodes and created a dictionary where a root-node key links to a list of all recursive children of that node in the graph. Only those terms with cross-referenced GO identifiers were included in the final mapping. The category subgraphs created from UniProt were compared to those with corresponding category root-nodes made by GOcats. An inclusion index, I, was calculated by considering the two subgraphs’ members as sets and applying the following equation:
(1) (Due to technical limitations, the equation has been placed in the Supplementary Files section.)
where Sn and Sg are the set of members within the non-GOcats-derived category and GOcats-derived category, respectively. It is worth noting here that the size of the UniProt set was always smaller than the GOcats set. This is due to the inherent size differences between UniProt’s CV and the Cellular Component sub-ontology.
Creating category mappings from Map2Slim
The Java implementation of OWLTools’ M2S does not include the ability to output a mapping file between fine-grained GO terms and their GO slim mapping target from the GAF that is mapped. To compare subgraph contents of GOcats categories to a comparable M2S “category,” we created a special custom GAF where the gene ID column and GO term annotation column of each line were each replaced by a different GO term for each GO term in Cellular Component, data-version: releases/2016-01-12. We then allowed M2S to map this GAF with a provided GO slim. The resulting mapped GAF was parsed to create a standalone mapping between the terms from the GO slim and a set of the terms in their subgraphs.
Mapping gene annotations to user-defined categories
To allow users to easily map gene annotations from fine-grained annotations to specified categories, we added functionality for accepting GAFs as input, mapping annotations within the GAF and outputting a mapped GAF into a user-specified results directory. The input-output scheme used by GOcats and M2S are similar, with the exception that GOcats accepts the mapping dictionary created from category keywords, as described previously, instead of a GO slim. GAFs are parsed as a tab-separated-value file. When a row contains a GO annotation in the mapping dictionary, the row is rewritten to replace the original fine-grained GO term with the corresponding category-defining GO term. If the gene annotation is not in the mapping dictionary, the row is not copied to the mapped GAF, and is added to a separate file containing a list of unmapped genes for review. The mapped GAF and list of unmapped genes are then saved to the user-specified results directory.
Visualizing and characterizing intersections of category subgraphs
To compare the contents of category subgraphs made by GOcats, UniProt CV, and M2S, we took the set of subgraph terms for each category in each method, converted them into a Pandas DataFrame (28) representation, and plotted the intersections using the UpSetR R package (24). Inclusion indices were also computed for M2S categories using Equation 1. Jaccard indices were computed for every subgraph pair to evaluate the similarity between subgraphs of the same concept, created by different methods.
Assigning generalized subcellular locations to genes from the knowledgebase and comparing assignments to experimentally-determined locations
We first mapped two GAFs downloaded from the EMBL-EBI QuickGO resource (12) using GOcats, the UniProt CV, and M2S. We filtered the gene annotations by dataset source and evidence type, resulting in separate GAFs containing annotations from the following sources: UniProt-Ensembl, and HPA. Both GAFs had the evidence type, inferred from Electronic Annotation, filtered out because it is generally considered to be the least reliable evidence type for gene annotation and in the interest of minimizing memory usage. We used this data to assess the performance of the mapping methods in their ability to assign genes to subcellular locations based on annotations from knowledgebases by comparing these assignments to those made experimentally in HPA’s localization dataset (Figure 3A). Comparison results for each gene were aggregated into 4 types: i) “complete agreement” for genes where all subcellular locations derived from the knowledgebase and the HPA dataset matched, ii) “partial agreement” for genes with at least one matching subcellular location, iii) “partial superset” for genes where knowledgebase subcellular locations are a superset of the HPA dataset, iv) "no agreement" for genes with no subcellular locations in common, and v) “no annotations” for genes in the experimental dataset that were not found in the knowledgebase.
Only gene product localizations from the HPA dataset with a “supportive” confidence score were used for this analysis (n=4795). We created a GO slim by looking up the corresponding GO term for each location in this dataset with the aid of QuickGO term basket and filtering tools. The resulting GO slim served as input for the creation of mapped GAFs using M2S. To create mapped GAFs using GOcats, we entered keywords related to each location in the HPA dataset (Table 4). We matched the identifier in the “gene name” column of the experimental data with the identifier in the “database object symbol” column in the GAF to compare gene annotations. Our assessment of comparing the HPA raw data to mapped gene annotations from the knowledgebase represents the ability to accurately query and mine genes and their annotations from the knowledgebase into categories of biological significance. Our assessment of comparing the methods’ mapping output to the HPA raw dataset represents the ability of these methods to evaluate the representation of HPA’s latest experimental data as it exists in public repositories.
Running time tests.
For comparing the runtimes of GOcats and M2S for categorizing HPA’s subcellular location dataset, each method was run separately on the same machine with the following configuration: Intel ® Core ™ i7-4930K CPU with 6 hyperthreaded cores clocked at 3.40GHzn and 64 GB of RAM clocked at 1866 MHz. We used the Linux “time” command with no additional options and reported the real time from its output. The datasets and scripts used can be found in our FigShare (See Availability of Data and Material). We used the dataset contained in our ScriptsDirectory/KBData/11-02-2016/hpa-no_IEA.goa for these comparisons. For M2S we executed a custom script that can be found within ScriptsDirectory/runscripts:
sh owlmultitest.sh
which ran the following command, found in the same subdirectory, 50 times:
time sh owltoolsspeedtest.sh
For GOcats, we executed a custom script that can be found within ScriptsDirectory/runscripts:
sh gcmultitest.sh
which ran the following command, found in the same subdirectory, 50 times:
time sh GOcatsspeedtest.sh
Both tests were executed using the same version of the go-core used across all other analyses performed in this work, which is data version: releases/2016-01-12.
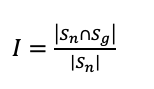{kind=link}