Antibodies and Reagents
The GST (0020), β-tubulin (0119), and IκBα (0040) antibodies were purchased from BioBharati LifeScience. The RelA (sc-372) antibody used in Western blots and ChIP assays was purchased from Santa Cruz Biotechnology. The FLAG (F1804) and control IgG ((12–371) antibodies were purchased from Sigma. The p84 (C1C3) antibody was purchased from GeneTex. The NME1 (3345) antibody was purchased from Cell Signaling. Mouse TNFα (BioBharati) was used at a final concentration of 20ng/mL for the indicated timepoints.
Mammalian Cell Culture and Transient Transfection
HeLa and HEK293T cell lines were maintained in Dulbecco’s modified Eagle’s medium (Corning) supplemented with 10% FBS and antibiotics. For transient transfection of HEK293T, cells were plated in 6-well dishes at 80% confluence the day before transfection. The next day, 2 µg of DNA was mixed with 8 µg of polyethylenimine (PEI) in OptiMEM (Gibco) at a total reaction volume of 50 µL. After incubation at room temperature for 15 minutes, cell media was exchanged to DMEM without FBS or antibiotics and DNA:PEI complexes were added dropwise over cells and left to incubate for 4 hours at 37°C. Media was then exchanged back to DMEM supplemented with 10% FBS and antibiotics and incubated for 16 hours before harvesting. For preparation of HeLa nuclear extract, plated cells were harvested by scraping in PBS and lysed in hypototonic lysis buffer consisting of PBS supplemented with 0.1% (v/v) NP-40, 1 mM DTT, and 0.25mM PMSF. Nuclei were pelleted by centrifugation at 3000g, washed twice with ice-cold PBS, and resuspended in nuclear extraction buffer containing 25 mM Tris-HCl pH 7.5, 420 mM NaCl, 10% glycerol, 0.2 mM EDTA, 1 mM DTT, 0.5 mM PMSF, and mammalian protease inhibitor cocktail (Sigma). After incubation on ice for 30 minutes, nuclei were centrifuged at 13,000 rpm for 15 minutes at 4°C. The supernatant containing nuclear extract was collected, snap frozen on liquid nitrogen, and stored at -80°C.
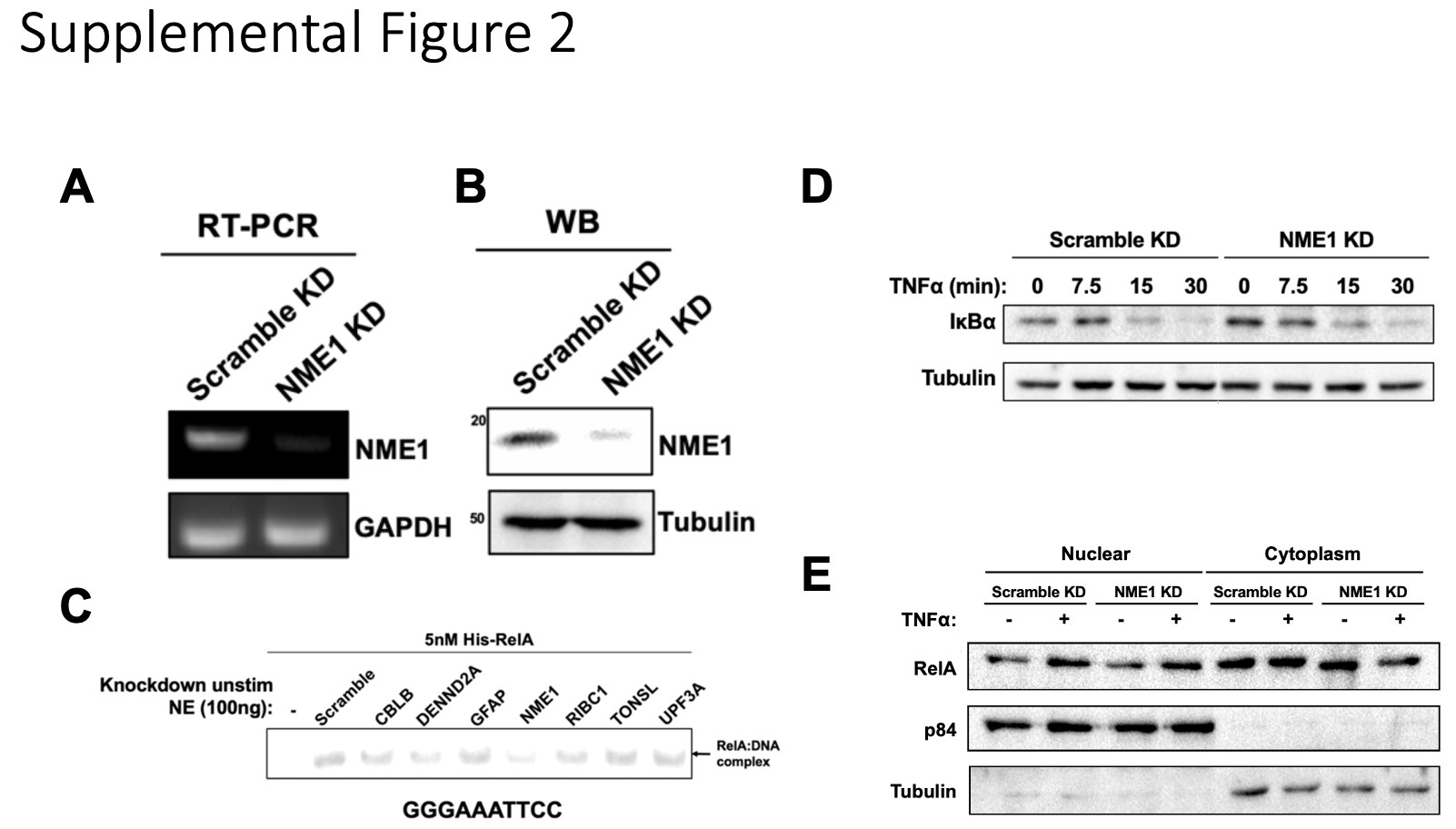
The list of oligonucleotides used for generation of stable knockdown HeLa cells are listed in Supplementary Table 1. First, forward and reverse oligonucleotides for specific targets were annealed in buffer containing 10 mM Tris-HCl pH 7.5, 50 mM NaCl, and 1 mM EDTA by incubating in boiling water allowed to slowly cool to room temperature. Annealed DNA duplex was then ligated into the pLKO.1 TRC vector using the AgeI and EcoRI restriction sites. Lentivirus was then generated in 293T cells by cotransfecting the pLKO.1 construct with pMDLg/pRRE, pCMV-VSV-G, and pRSV-Rev expression constructs. After 48 hours, virus-containing supernatant was harvested, filtered through a 0.4 µm filter, and used to infect HeLa at a 1:10 dilution in the presence of 10 ng/µL polybrene (MilliporeSigma). After 48 hours, stably infected cells were selected by treatment of 1 µg/mL puromycin in culture media and continuously maintained in the presence of 1 µg/mL puromycin.
Protein Expression and Purification
Expression and purification of His-tagged full-length RelA from Sf9 cells and His-tagged RelA RHR (19–304) in E. coli Rosetta (DE3) was performed as previously described (Li et al., 2024).
His-tagged NME1 was cloned into the pET-24d vector and expressed in E. coli Rosetta (DE3) by growing cells to an OD600 of 0.4 and inducing with 0.25 mM IPTG overnight at room temperature. Cells were pelleted by centrifugation and lysed by sonication in lysis buffer containing 25 mM Tris-HCl pH 7.5, 300 mM NaCl, 5% glycerol, 0.1% NP-40, 10 mM imidazole, 5 mM β-mercaptoethanol, and 0.25 mM PMSF. Lysate was clarified by centrifugation at 14,000 rpm for 30 minutes at 4°C and incubated with nickel-NTA agarose beads (BioBharati LifeScience) for 2 hours at 4°C on a rotary shaker. Beads were then extensively washed with lysis buffer and His-tagged NME1 was eluted in lysis buffer without PMSF but supplemented with 250 mM imidazole. Protein quality was assessed by SDS-PAGE with Coomassie staining and peak fractions were pooled, snap frozen on liquid nitrogen, and stored at -80°C.
NME1 was cloned into the pGEX-4T2 GST vector and both GST and GST-tagged NME1 were expressed in E. coli Rosetta (DE3). Cells were grown to an OD600 of 0.3 and induced with 0.25 mM IPTG overnight at 16°C. Cells were then pelleted by centrifugation and lysed by sonication in lysis buffer containing 50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol, 0.1% NP-40, 0.5 mM EDTA, 1 mM DTT, and 0.25 mM PMSF. The lysate was then clarified by centrifugation at 14,000 rpm for 30 minutes at 4°C and incubated with pre- equilibrated glutathione agarose beads (BioBharati LifeScience) for 2 hours at 4°C on a rotary shaker. Beads were then extensively washed with lysis buffer and eluted lysis buffer without PMSF but with 25 mM reduced glutathione (Sigma-Aldrich). Eluted protein was then dialyzed in a 6–8 kDa MWCO membrane three times in 1L of dialysis buffer containing 50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5% glycerol, 0.5 mM EDTA, and 1 mM DTT and dialyzed once in 200 mL of dialysis buffer containing 50% glycerol. The dialyzed 50% glycerol stock protein was then stored at -20°C.
Electrophoretic Mobility Shift Assay
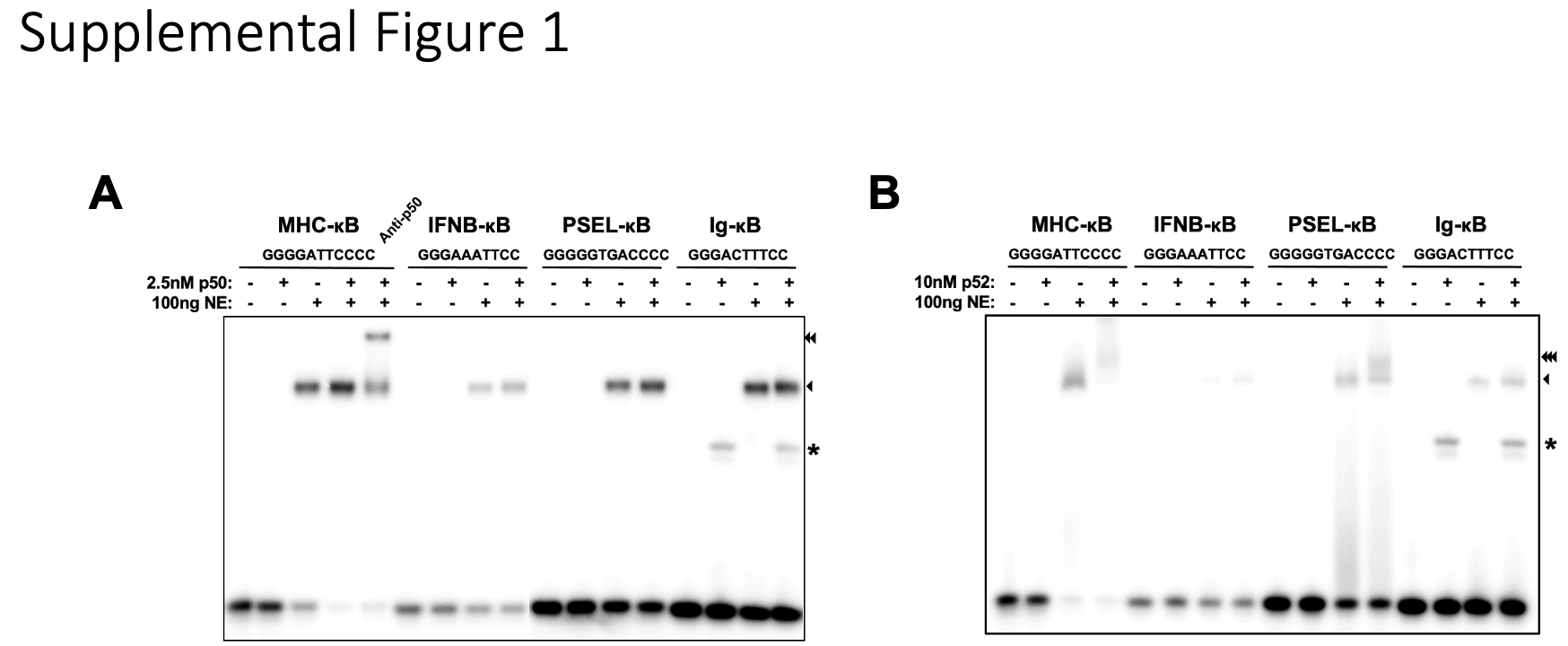
Radiolabeled probes were incubated with the proteins under study for 20 minutes at room temperature in binding buffer containing 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol, 1% NP-40, 1 mM EDTA, and 0.1 mg/mL poly(dI-dC). When needed, proteins were diluted in dilution buffer containing 20 mM Tris- HCl pH 7.5, 50 mM NaCl, 10% glycerol, 1 mM DTT, and 0.2 mg/mL BSA. Samples were run through a 4% nondenatured polyacrylamide gel in TGE buffer (24.8 mM Tris base, 190 mM glycine, and 1 mM EDTA) at 200V for 1 hour. Gel was then dried, exposed on a phosphor screen overnight, and scanned by Typhoon FLA 9000 imager (Cytiva).
Fractionation and Identification of RelA-Specific Cofactors
Unstimulated HeLa nuclear extract was collected from three 15 cm dishes in a total volume of 500 µL and cleared by centrifugation at 13,000 rpm for 15 minutes at 4°C. Supernatant was collected and fractionated through a pre-equilibrated 24 mL Superose 6 (Cytiva) size-exclusion column in SEC buffer containing 25 mM Tris-HCl pH 7.5, 420 mM NaCl, 10% glycerol, 0.2 mM EDTA, 1 mM DTT, and 0.5 mM PMSF. 42 fractions at a volume of 330 µL (14 mL total volume) were collected starting 8 mL after injection, and 2 µL from fractions were tested for activity by EMSA with 5 nM of recombinant full-length RelA. Fractions 16–19 showed the most activity and were therefore combined and diluted in SEC buffer without NaCl to a final 100 mM NaCl concentration. The pooled extract was then centrifuged at 13,000 rpm for 15 minutes at 4°C and supernatant was passed through a 3 mL Mono Q (Cytiva) anion exchange column. The column was then washed with 10 mL of buffer containing 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 10% glycerol, and 1 mM DTT and bound proteins were eluted with 10 mL of the same buffer but with an increasing NaCl gradient up to 700 mM NaCl. A total of 40 250 µL fractions were collected and 2 µL from fractions were tested for activity by EMSA with recombinant full-length RelA.
The fractions corresponding to Q20, Q23, Q26, Q29, and Q32 showed the highest activity and were further tested for RelA DNA-binding enhancement in an in vitro biotinylated DNA-pulldown assay. Biotinylated DNA was annealed as outlined previously in buffer containing 10 mM Tris-HCl pH 7.5, 50 mM NaCl, and 1 mM EDTA and immobilized onto streptavidin agarose beads (BioBharati LifeSciences) in buffer containing 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, 0.1% and 1 mM DTT. Beads were then washed to remove unbound DNA and mixed with 100 ng of recombinant full-length RelA at a total volume of 200 µL. 10 µL of the corresponding Mono Q fractions were added and samples were rotated for 2 hours at 4°C. Beads were then pelleted by centrifugation and washed 4 times with the same pulldown buffer. After the final wash, 4x SDS gel loading dye was added to the beads at a final dilution of 1x and beads were boiled for 10 minutes. Samples were centrifuged at 13,000 rpm for 5 minutes and supernatant was separated by SDS-PAGE and analyzed by Western blot.
Fraction Q23 showed the highest activity in both EMSA and the DNA pulldown assay and was therefore further investigated by mass-spec for peptide identification. A final pulldown was performed in
buffer containing 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, and 1 mM DTT with 100 ng of recombinant full-length RelA, fraction Q23, and biotinylated-DNA immobilized onto streptavidin agarose beads. A control pulldown was also prepared in parallel without recombinant RelA added. The reactions were incubated overnight at 4°C with gentle rotation. Beads were then washed with the pulldown buffer 3 times and precipitated peptides were identified by liquid chromatography with mass spectrometry (LC- MS) at the UCSD Biomolecular and Proteomics Mass Spectrometry Facility.
Luciferase Assays
Complementary oligonucleotides were first annealed by mixing at a final concentration of 2 µM in buffer containing 10 mM Tris-HCl pH 7.5, 50 mM NaCl, and 1 mM EDTA and incubating in boiling water allowed to slowly cool to room temperature. Annealed promoters were then cloned into the CMXTK- Luciferase vector (a kind gift from Dr. Chakravarti at Northwestern University Feinberg School of Medicine) at the SalI and BamHI restriction sites. HEK293T were grown in 12-well plates and transiently transfected with the FLAG-tagged overexpression construct or empty vector control, luciferase reporter DNA, and control CMV- driven Renilla. After 24 hours, cells were stimulated for 8 hours with 20 ng/mL mouse TNFα. Lysate was then collected and used for luciferase activity assay using the Dual-Luciferase Reporter Assay System (Promega). Data are represented as mean ± standard deviations (SD) of three or more independent experimental replicates.
Pulldown Assays
NME1 was cloned into a modified pEYFP-c1 vector with YFP removed and substituted for an N-terminal FLAG tag. The day before transfection, HEK293T was plated on a 6-well plate. The next day, the cells were at approximately 80% confluence and transfected with either FLAG empty vector or FLAG-tagged NME1 with PEI. The next day, cells were washed with ice-cold PBS and lysed directly on the plate in buffer containing 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, 1% NP-40, 1 mM DTT, and 0.5 mM PMSF by gentle rocking at 4°C for 15 minutes. Lysate was then collected and centrifuged at 13,000 rpm for 15 minutes at 4°C. Supernatant was then collected and mixed with 50 µL of preequilibrated anti- FLAG M2 beads (Sigma) for 2 hours at 4°C on a rotary shaker. Beads were then extensively washed and mixed with 4x SDS gel loading dye to a final dilution of 1x. Samples were boiled for 5 minutes and centrifuged for 5 minutes at 13,000rpm. Supernatant was then resolved by SDS-PAGE and analyzed by Western blot.
For GST pulldown assays, first 1µg of GST or GST-tagged NME1 was mixed with 50 µL of preequilibrated glutathione agarose beads (BioBharati LifeScience) in 200 µL of pulldown buffer containing 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, 0.1% NP-40, and 1 mM DTT. Samples were mixed for 1 hour at 4°C with gentle rotation. Beads were then washed 4 times with pulldown buffer to wash unbound proteins and brought to a final volume of 200 µL after the last wash. 1 µg of recombinant full-length RelA and 1 µM of annealed κB or mutant κB DNA was then added and the reaction was incubated for 2 hours at 4°C with gentle rotation. Beads were then extensively washed and mixed with 4x SDS gel loading to a final dilution of 1x. Samples were then boiled for 5 minutes and
centrifuged at 13,000rpm for 5 minutes. Supernatant was then resolved by SDS-PAGE and analyzed by Western blot.
RNA Isolation and Real-Time qPCR
A list of oligonucleotides used in RT-qPCR reactions is listed in Supplementary Table 1. HeLa was plated in 6 well dishes and total RNA was isolated the next day with TRIzol (Invitrogen) and purified by isopropanol precipitation following the manufacturers recommendations. RNA concentration was determined by nanodrop and cDNA was synthesized in a 5 µL reaction from 500 ng of RNA using SuperScript IV VILO Master Mix (ThermoFisher). The cDNA was then diluted 1:4 to a total volume of 20 µL and 1 µL was used as template for qPCR with the Luna qPCR Master Mix (New England Biolabs) in a total reaction volume of 10 µL. Values were normalized to GAPDH and data are represented as mean ± standard deviation of three independent experimental replicates.
Chromatin Immunoprecipitation qPCR
A list of oligonucleotides used in ChIP-qPCR experiments is listed in Supplementary Table 1. For every two immunoprecipitation reactions, a confluent 10 cm dish of HeLa was used. Cells were first treated with TNFα or DMSO for 30 minutes. Formaldehyde was then added directly to the media at a final concentration of 1% and incubated on the cells for 10 minutes at room temperature with gentle rocking. Crosslinking was then quenched by addition of 125 mM glycine and incubation at room temperature for 5 minutes with gentle rocking. Cells were then washed twice with ice-cold PBS and collected by scraping in 1mL of PBS. Cells were then pelleted by centrifugation and resuspended in 1 mL of PBS supplemented with 0.1% NP-40, 1 mM DTT, and 0.25 mM PMSF. The cell pellet was then gently pipetted 5 times to facilitate cytoplasmic lysis and nuclear fractionation, and nuclei was then pelleted by centrifugation. The nuclear pellet was then resuspended in 1 mL of RIPA buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 0.5 mM PMSF, and mammalian protease inhibitor cocktail (Sigma). Resuspended nuclei were sonicated on ice with a micro tip sonicator (Branson) to generate DNA fragments with an average length of 500 bp. Sonicated nuclear extract was then centrifuged at 13,000 rpm for 15 minutes at 4°C and supernatant was precleared with 25 µL protein AG PLUS agarose beads (BioBharati LifeScience) and 500 ng of IgG control antibody (ThermoFisher) for 1 hour at 4°C with rotation. Extract was then centrifuged at 13,000 rpm for 10 minutes at 4°C, and precleared supernatant was divided into two equal reactions and mixed with 25 µL of protein AG PLUS agarose beads and 500 ng of either IgG or anti-RelA antibody. Immunoprecipitation reactions were incubated at 4°C overnight with gentle rotation. Beads were then washed three times for 5 minutes each with 1 mL of RIPA buffer, then RIPA buffer supplemented with 500 mM NaCl, RIPA buffer with NaCl substituted with 250 mM LiCl, and lastly with TE buffer containing 10 mM Tris-HCl pH 7.5 and 1 mM EDTA. Immune complexes were then eluted from beads in 150 µL of elution buffer containing 1% SDS and 100 mM sodium bicarbonate pH 8.0 for 30 minutes at room temperature with rotation. Eluted complex was then mixed to 6 µL of 5 M NaCl and 2 µL of 100 mg/mL RNase A (Qiagen) and incubated overnight at 65°C to reverse cross-linking and digest RNA. The next morning, 2 µL of Proteinase K (Invitrogen) was added and incubated at 60°C for 1 hour. DNA was then extracted with phenol:chloroform:isoamyl alcohol (25:24:1) (Invitrogen) and isopropanol precipitation following manufacturers recommendation. DNA pellet was resuspended in 50 µL of TE buffer and 1 µL was used in qPCR reaction with Luna qPCR Master Mix (New England Biolabs).
RNA Sequencing and Analysis
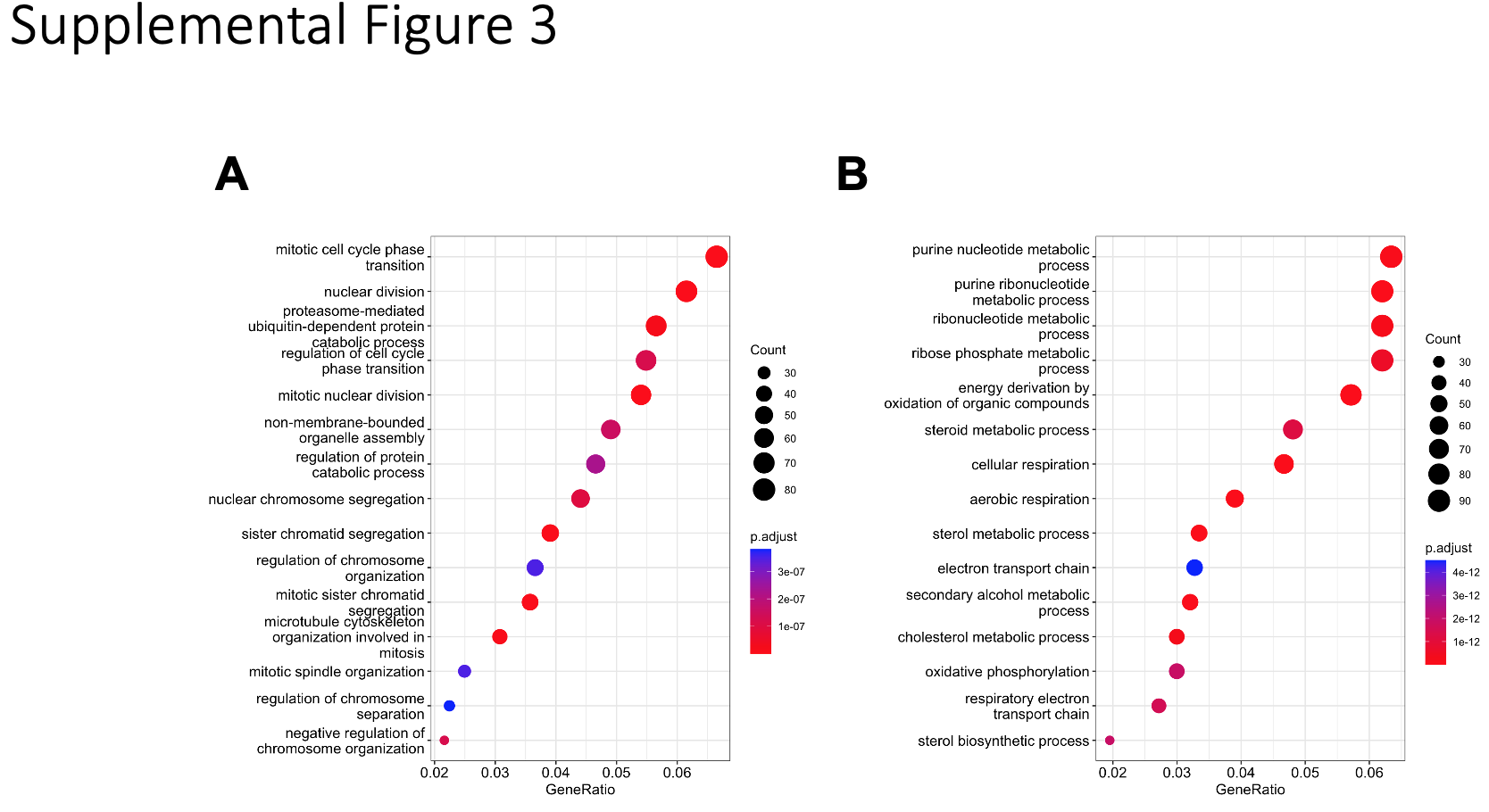
Scramble and NME1 knockdown HeLa cell lines were initially plated on 6 well plates. The next day, duplicate wells from each cell line were treated for 1 hour with either DMSO or 20 ng/mL TNFα. Cells were then washed once with ice-cold PBS and RNA was isolated with TRIzol (Invitrogen) following manufacturers recommendations. RNA quality was then assessed by TapeStation (Agilent) and RNA with a RIN score greater than 8.0 was further processed. Poly-A enriched libraries were prepared from 1 µg of total RNA using the mRNA HyperPrep Kit (KAPA) with unique dual-indexed adapters (KAPA) following manufacturers recommendations. Library quality was assessed by DNA TapeStation (Agilent) and quantified with Qubit 2.0 fluorometer (Life Technologies). Libraries were pooled and underwent paired-end sequencing using the NovaSeq6000 (Illumina) at the UCSD Institute for Genomic Medicine (IGM). For analysis of sequencing data, read quality was first checked by FASTQC. Reads were then mapped to human genome using OSA/Oshell (Omicsoft). Reads were then normalized and differentially expressed genes were analyzed using DESeq2 (v1.38.3).
{kind=link}
{kind=link}
{kind=link}