The field of computational drug design is rapidly advancing, necessitating innovative methods to enhance the efficiency and accuracy of ligand-receptor interactions. We introduce M01 tool, a comprehensive computational package designed to facilitate the generation and docking of small molecule-peptide hybrids. M01 tool integrates several established tools, including RDKit and EasyDock, into a user-friendly platform that automates the workflow from hybrid generation to docking simulations. Key features include an intuitive interface for visualizing molecules and selecting connection points, automated receptor preparation from UniProt or PDB IDs, generation of default docking configuration files, ligand preparation and docking using EasyDock, and calculation of molecular descriptors related to ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties. M01 tool aims to simplify the use of advanced computational tools for researchers with limited chemistry expertise, providing a robust and streamlined solution for hybrid design and docking studies. Validation using peptide-alkoxyamine hybrids demonstrated M01 tool's capability to generate and dock over 8,000 unique hybrid molecules, confirming its potential as a powerful tool in drug design.
Method Article
M01 tool: An automated, comprehensive computational tool for generating small molecule-peptide hybrids and docking them into curated protein structures
https://doi.org/10.21203/rs.3.rs-5248397/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Computational Drug Design
Hybrid Molecules
Docking Simulations
Ligand-Receptor Interactions
ADMET Properties
The field of computational drug design is a rapidly evolving area that plays a crucial role in the identification and optimization of potential drug candidates. A majority of compounds undergoing drug development procedures fail, consequently need structural alteration to yield better efficacy, pharmacokinetic properties, and effectiveness (Sun, Gao et al. 2022). Despite extensive efforts and resources spent on the matter, the cost and time to introduce novel drug candidates have increased over the last three decades (Kiriiri, Njogu et al. 2020). Conventional computational methods, including docking simulations, have significantly enhanced the efficiency and accuracy of ligand-receptor interaction studies (Bhagat, Butle et al. 2021). Yet, these methods often require specialized expertise and involve complex procedures, creating a barrier for researchers who lack a strong background in chemistry. Thus, developing integrable, automated and easily applicable softwares seems attractive to researchers.
One previously created semi-automated docking tool is EasyDock (Minibaeva, Ivanova et al. 2023), designed by Minibaeva et al. for customizable docking with proper ligand preparation and optimization for further docking processes. It primarily uses RDKit modules for three dimensional (3D) embedding, substituting boron atoms, in case found in the ligand structure, with carbons for achieving the possibility of docking calculations through Vina, and creating a PDBQT file via the Meeko module. It also enables users to protonate ligands with the pkasovler toolkit, which predicts protonation state with graph convolutional networks. In addition to EasyDock, there are also other necessary modules for docking processes, including PDBFixer for receptor file curation and MGLTools for PDBQT file creation, which need to be pipe-lined to be used effortlessly. Besides, the ability to generate complex-structured ligands can facilitate enhanced ligand-receptor interactions.
Hybrid compounds consisted of small molecule and peptide components are of attractive complex molecules which have emerged as promising candidates in drug design (Wang, Yang et al. 2019, Feng, Zheng et al. 2021, Wang, Tsuji et al. 2022). Such hybrids may combine the favorable properties of both endogenous peptide ligands and small-molecule antagonists of the corresponding receptor. Peptides themselves have been shown to have a key role in regulating many biological processes, including pathological states of pain (Nemoto, Yamagata et al. 2023) and multiple behavioral processes (Bhat, Shahi et al. 2021, Shevchouk, Tufvesson-Alm et al. 2021, Nemoto, Yamagata et al. 2023), as well as showing antibiotic properties (Massari, Nannetti et al. 2015). Hybridizing molecules can offer numerous advantages, including overcoming drug resistance (Ouji, Nguyen et al. 2021), improving the solubility of other drugs (Liu, Zhao et al. 2023), modulating multiple targets (Dumitrascuta, Bermudez et al. 2021). Thus, rational binding of peptides to small molecules can exhibit enhanced properties. In this article, we refer to the small-molecule substructure of the hybrid as “core ligand” and to the amino acids and/or other hybridizing molecules as “hybridant”. Despite the aforementioned potentials, there are numerous problems intrinsic to small oligopeptides themselves, such as biodegradation, increased clearance, and poor bioavailability (Song, Fan et al. 2021). Pseudopeptides are generated to address such issues (Mäde, Els-Heindl et al. 2014, Sun, Li et al. 2017). This includes substituting L-amino acids with D-types, changing amide bonds, cyclization of the peptides, etc.(Lee 2022).
To address these challenges, we developed M01 tool, a fully automated computational package for generating small molecule-peptide hybrids then docking them into a curated protein structure in a comprehensive pipe-lined module. M01 tool integrates several established tools, including RDKit, EasyDock, PDBFixer, MGLTools, and Autodock Vina (Trott and Olson 2010, Eberhardt, Santos-Martins et al. 2021) into a cohesive platform that streamlines the workflow from hybrid generation to docking simulation. This tool is designed to be functional to researchers with limited chemistry expertise. In comparison with existing tools, M01 tool offers full automation through receptor download, preparation, and curation options. Receptor curation is modulated with PDBFixer package, which searches the protein structure for missing or non-standard residues and atoms and replaces them according to SEQRES records which contain the sequences of the reported protein tertiary structures. Then a default docking configuration is considered, minimizing user interaction and docking expertise. In addition, we provide users with a user-friendly graphic user interface (GUI) as well as molecular descriptor calculations for pharmacokinetic and ADMET predictions, paving the way for further analysis and predictions.
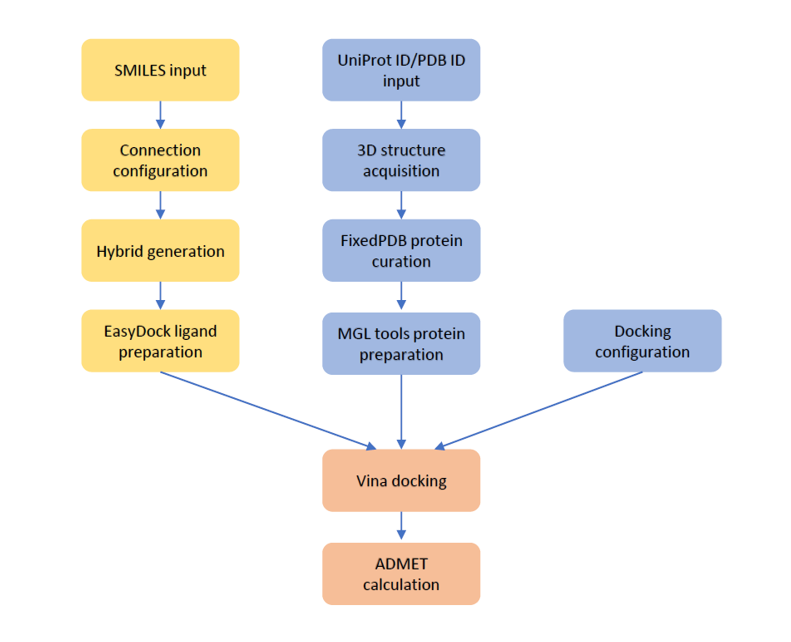
The workflow designed in M01 tool software is illustrated in Scheme 1.
[Scheme 1 near here]
2.1. Hybrid Generation
The hybrid generation method involves creating small molecule-peptide hybrids using the RDKit module. The RDKit library for molecular manipulation and hybridizing small molecules with peptides was used. The core component of this method is the LigandBuilder class, which handles the creation of peptide bonds and the removal of the hydrogens needed for the peptide bond formation. A view of the graphical online tool is demonstrated in scheme 2.
[Scheme 2 near here]
- The input_amino_acids function allows users to input sequences of L- and D-amino acids, as well as other molecules, in various combinations. These sequences are then converted into RDKit molecule objects. The “other molecules” option is provided to handle non-standard amino acids as well as bulky and protective groups that possess the capability of forming at least one peptide bond from the selected terminal.
- The create_peptide_bond method creates peptide bonds between pairs of molecules, ensuring that the combined molecule retains a valid structure. Four possible reactions are attempted to maximize the success rate of bond formation in the molecules containing carboxylic acid or carbonate substructures.
- The remove_extra_hydrogens method maintains the structural integrity of the hybrid molecules and removes the extra hydrogen atoms selectively from the nitrogen atoms. Special care is taken for aromatic heterocycle structures, ensuring that the nitrogen atoms within these structures are properly hydrogenated. These structures can lead to kekulization errors if not handled properly.
- The generate_ligands method generates all possible ligand arrangements by combining the input amino acids with a specified molecule. The connection site and terminal (C or N) are specified to guide the attachment process. The process generates unique ligands by attaching peptide sequences to the specified connection site of the input small molecule. Each generated ligand is then checked for chemical accuracy, hydrogenated, and converted to its canonical SMILES representation for further use in docking simulations.
2.2. Ligand preparation
Ligand preparation is implemented via the EasyDock module, to which Ligands can be provided as SMILES strings. If non-3D structures are provided, RDKit’s EmbedMolecule and UFFOptimizeMolecule modules will create optimized structures. Protonation is implemented at pH 7.4 or any desired pH with the pkasolver module. Although automated protonation applications are not optimal (ten Brink and Exner 2009), their use in high-throughput practices is widely accepted (Bender, Gahbauer et al. 2021). Then, the molecule is converted to PDBQT format via the Meeko module and can be used for docking simulation.
2.3. Target preparation
Either UniProt (2023) or PDB IDs (Berman, Westbrook et al. 2000) can be used as input to prepare a receptor ready for further docking calculations. In the event of receiving a UniProt ID, the program connects to the UniProt API (application programming interface) provided by the database to query and access its data programmatically. Data retrieved from the API is then parsed to search for PDB IDs and chains related to the desired protein. In cases of multiple PDBs per entry, the chain with the largest amino acid count and highest resolution is selected. After the PDB ID and chain are chosen, the PDB file is downloaded with the Biopython module (Cock, Antao et al. 2009), and extra chains are removed from the file. In this step, the centroid of the chain is also calculated and added to the PDB file in order to use in further docking calculations. The PDBFixer module is also integrated with the target preparation module, which is used to fix common problems in PDB files, such as missing atoms, missing residues, non-standard residues, etc. Additionally, all heteroatoms, including water molecules, are removed in this step. PDB files with multiple chains are accessible through PDB IDs.In M01 TOOL, the MGLtools tool was used to check hydrogens, add Gasteiger charges (Tiwari, Mahasenan et al. 2009) to the structure and remove non-polar hydrogens by merging them into the adjacent carbon atom.
2.4. Docking configuration
This tool automatically generates default configuration files for docking, thus, minimizing the needed input files. The latest version of Autodock Vina (1.2.5) was used for docking simulations. The input PDBQT files and chain centroid are prepared as explained in the previous sections, while the protein setup and config files, each one file, are yet to be prepared. The chain centroid is used as the center of the grid box and a box of 126×126×126 Å is selected to cover the whole chain in order to run a blind docking process. The high value of 32 is chosen for exhaustiveness to compensate for a fairly large grid box size (Trott and Olson 2010, Eberhardt, Santos-Martins et al. 2021). Maximally 10 binding modes per ligand will be obtained. In the subsequent analysis the highest binding affinity will only be considered.
2.5. Molecular descriptor calculation
For each ligand, M01 tool calculates the Crippen-Wildman partition coefficients (Wildman and Crippen 1999) (logP), hydrogen bond donor count, hydrogen bond acceptor count, molecular weight (Lipinski 2000), topological polar surface area (TPSA), and QED score (Bickerton, Paolini et al. 2012). This calculation is performed through RDKit’s rdMolDescriptors module.
In this section we evaluate the performance of the tool with regenerating a set of previously generated ligands and checking the validity of the generated molecules.
Performance
For confirming accurate performance of M01 tool software in hybrid structure generation a set of peptide-alkoxyamine hybrids were used in validation step. They were previously studied for their antiplasmodial activity by Embo-Ibouanga et al (Embo-Ibouanga, Nguyen et al. 2024). The validation process involved selecting the core molecule, selecting the hybridants and searching for the expected hybrids between the results. Hybrids were generated using three D-amino acids (D-valine, D-phenylalanine and D-valine) and three L-amino acids (L-valine, L-phenylalanine and L-valine). These amino acids were inputted in FASTA format. The hybrid generation process yielded 8,163 unique hybrid molecules, consisting of all possible combinations of hybridants, all of which were successfully docked into the receptor. Generated hybrids were consisted of all the molecules tested in the study by Embo-Ibouanga et al and new hybrid combinations. A list of amino acids and other molecules used for this hybridization are provided in Table 1.
Table 1: List of Amino Acids and Other Molecules Used for Hybridization

Limitations and outlook
The model's performance may be constrained by the computational power available, particularly for large-scale docking simulations of complex molecular structures. The number of possible structures and the size of the grid box are considerably high and require a lot of computational power. However, even the largest grid box fails to contain all molecules of large protein structures necessitating a more precise, active-site-based grid center. In addition, the use of UniProt IDs as input can pose certain limitations on the user since each UniProt entry contains only one subunit of each protein, while many proteins have multiple peptide or nucleic acid chains. Certain ligands, such as fluoroquinolone antimicrobials, connect to the cleavage site somewhere between the two protein chains of their target receptor, DNA gyrase, and interact with the DNA as well. In such cases, using the uniport ID cannot yield acceptable results.
With the introduction of M01 tool, we aim to automate the docking process extensively and intend to make it convenient to use by all the scientific community for the implementation of complex de novo drug design models. The options of docking with nucleic acid, web server platform, homology modelling of proteins, and customizable grid boxes are left for future works. Additionally, user feedback and real-world applications will provide valuable insights for continuous improvements.
This study presents an automated hybrid generating and docking pipeline, named M01 tool, in which the user can measure ligand-receptor affinities with minimal previous expertise in the field. This study is mainly consisted of previously generated modules that does not necessitate further validation. With the advent of artificial intelligence and especially machine learning methods, there appears to be an increasing need for simple platforms that can provide models with a robust, easy-to-implement reward system for new molecules, which can be handled with this automated customizable python-based docking package. The hybrid generating tool is available at: https://m01tool.com/ .
Availability and requirements
Project name: M01 tool.
Project home page: https://github.com/mahsasheikh/M01-tool
Operating system(s): Platform independent.
Programming language: Python 3.
Other requirements: RDKit, vina, PDBFixer, mgltools.
License: MIT-license.
Any restrictions to use by non-academics: no limitation.
Supplementary Information
Author contributions
M.S. developed the program, performed tests and wrote the manuscript. M.N. developed
the program, developed the online tool. A.F. wrote the manuscript. All authors reviewed the manuscript.
Funding
No special funding was applied for this research.
Competing interests
The authors declare no competing interests.
- . "Meeko: preparation of small molecules for AutoDock." 2024, from https://github.com/forlilab/Meeko.
- . "MGLTools." 2024, from https://ccsb.scripps.edu/mgltools.
- . "PDBFixer." 2024, from https://github.com/openmm/pdbfixer.
- . "pkasolver." 2024, from https://github.com/mayrf/pkasolver.
- . "RDKit." 2024, from https://www.rdkit.org/.
- "UniProt: the universal protein knowledgebase in 2023." Nucleic acids research51(D1): D523-D531.
- Bender, B. J., S. Gahbauer, A. Luttens, J. Lyu, C. M. Webb, R. M. Stein, E. A. Fink, T. E. Balius, J. Carlsson and J. J. Irwin (2021). "A practical guide to large-scale docking." Nature protocols16(10): 4799-4832.
- Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne (2000). "The protein data bank." Nucleic acids research28(1): 235-242.
- Bhagat, R. T., S. R. Butle, D. S. Khobragade, S. B. Wankhede, C. C. Prasad, D. S. Mahure and A. V. Armarkar (2021). "Molecular docking in drug discovery." Journal of Pharmaceutical Research International33(30B): 46-58.
- Bhat, U. S., N. Shahi, S. Surendran and K. Babu (2021). "Neuropeptides and behaviors: how small peptides regulate nervous system function and behavioral outputs." Frontiers in Molecular Neuroscience14: 786471.
- Bickerton, G. R., G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins (2012). "Quantifying the chemical beauty of drugs." Nature chemistry4(2): 90-98.
- Cock, P. J., T. Antao, J. T. Chang, B. A. Chapman, C. J. Cox, A. Dalke, I. Friedberg, T. Hamelryck, F. Kauff and B. Wilczynski (2009). "Biopython: freely available Python tools for computational molecular biology and bioinformatics." Bioinformatics25(11): 1422.
- Dumitrascuta, M., M. Bermudez, O. Trovato, J. De Neve, S. Ballet, G. Wolber and M. Spetea (2021). "Antinociceptive efficacy of the µ-opioid/nociceptin peptide-based hybrid KGNOP1 in inflammatory pain without rewarding effects in mice: An experimental assessment and molecular docking." Molecules26(11): 3267.
- Eberhardt, J., D. Santos-Martins, A. F. Tillack and S. Forli (2021). "AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings." Journal of chemical information and modeling61(8): 3891-3898.
- Embo-Ibouanga, A. W., M. Nguyen, L. Paloque, M. Coustets, J.-P. Joly, J.-M. Augereau, N. Vanthuyne, R. Bikanga, N. Coquin and A. Robert (2024). "Hybrid Peptide-Alkoxyamine Drugs: A Strategy for the Development of a New Family of Antiplasmodial Drugs." Molecules29(6): 1397.
- Feng, L. S., M. J. Zheng, F. Zhao and D. Liu (2021). "1, 2, 3‐Triazole hybrids with anti‐HIV‐1 activity." Archiv der Pharmazie354(1): 2000163.
- Kiriiri, G. K., P. M. Njogu and A. N. Mwangi (2020). "Exploring different approaches to improve the success of drug discovery and development projects: a review." Future Journal of Pharmaceutical Sciences6(1): 27.
- Lee, Y. S. (2022). "Peptidomimetics and their applications for opioid peptide drug discovery." Biomolecules12(9): 1241.
- Lipinski, C. A. (2000). "Drug-like properties and the causes of poor solubility and poor permeability." Journal of pharmacological and toxicological methods44(1): 235-249.
- Liu, X., L. Zhao, B. Wu and F. Chen (2023). "Improving solubility of poorly water-soluble drugs by protein-based strategy: A review." International journal of pharmaceutics634: 122704.
- Mäde, V., S. Els-Heindl and A. G. Beck-Sickinger (2014). "Automated solid-phase peptide synthesis to obtain therapeutic peptides." Beilstein journal of organic chemistry10(1): 1197-1212.
- Massari, S., G. Nannetti, J. Desantis, G. Muratore, S. Sabatini, G. Manfroni, B. Mercorelli, V. Cecchetti, G. Palù and G. Cruciani (2015). "A broad anti-influenza hybrid small molecule that potently disrupts the interaction of polymerase acidic protein–basic protein 1 (PA-PB1) subunits." Journal of Medicinal Chemistry58(9): 3830-3842.
- Minibaeva, G., A. Ivanova and P. Polishchuk (2023). "EasyDock: customizable and scalable docking tool." Journal of Cheminformatics15(1): 102.
- Nemoto, W., R. Yamagata, O. Nakagawasai and K. Tan-No (2023). "Angiotensin-related peptides and their role in pain regulation." Biology12(5): 755.
- Ouji, M., M. Nguyen, R. Mustière, T. Jimenez, J.-M. Augereau, F. Benoit-Vical and C. Deraeve (2021). "Novel molecule combinations and corresponding hybrids targeting artemisinin-resistant Plasmodium falciparum parasites." Bioorganic & Medicinal Chemistry Letters39: 127884.
- Shevchouk, O. T., M. Tufvesson-Alm and E. Jerlhag (2021). "An overview of appetite-regulatory peptides in addiction processes; from bench to bed side." Frontiers in Neuroscience15: 774050.
- Song, H. Q., Y. Fan, Y. Hu, G. Cheng and F. J. Xu (2021). "Polysaccharide–peptide conjugates: a versatile material platform for biomedical applications." Advanced Functional Materials31(6): 2005978.
- Sun, D., W. Gao, H. Hu and S. Zhou (2022). "Why 90% of clinical drug development fails and how to improve it?" Acta Pharmaceutica Sinica B12(7): 3049-3062.
- Sun, X., Y. Li, T. Liu, Z. Li, X. Zhang and X. Chen (2017). "Peptide-based imaging agents for cancer detection." Advanced drug delivery reviews110: 38-51.
- ten Brink, T. and T. E. Exner (2009). "Influence of protonation, tautomeric, and stereoisomeric states on protein− ligand docking results." Journal of chemical information and modeling49(6): 1535-1546.
- Tiwari, R., K. Mahasenan, R. Pavlovicz, C. Li and W. Tjarks (2009). "Carborane clusters in computational drug design: a comparative docking evaluation using AutoDock, FlexX, Glide, and Surflex." Journal of chemical information and modeling49(6): 1581-1589.
- Trott, O. and A. J. Olson (2010). "AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading." Journal of computational chemistry31(2): 455-461.
- Wang, C., C. Yang, Y.-c. Chen, L. Ma and K. Huang (2019). "Rational design of hybrid peptides: a novel drug design approach." Current medical science39(3): 349-355.
- Wang, R., K. Tsuji, T. Kobayakawa, Y. Liu, K. Yoshimura, S. Matsushita, S. Harada and H. Tamamura (2022). "Hybrids of small CD4 mimics and gp41-related peptides as dual-target HIV entry inhibitors." Bioorganic & Medicinal Chemistry76: 117083.
- Wildman, S. A. and G. M. Crippen (1999). "Prediction of physicochemical parameters by atomic contributions." Journal of chemical information and computer sciences39(5): 868-873.
Schemes 1-2 are available in the Supplementary Files section.
The authors declare no competing interests.
- Scheme1.WorkflowofhybridgenerationanddockingusingM01tool.png
Workflow of hybrid generation and docking using M01 tool
- Scheme2.AviewoftheonlineM01tool.png
A view of the online M01 tool
{kind=link}
{kind=link}