Identification of circRNAs in GCs of SYFs in hens
Six circRNA libraries were built from RL (n = 3) and WL (n = 3) groups, an Illumina Hiseq 4000 instrument was applied to sequence these samples, and the obtained reads were mapped to the chicken reference genome (Gallus-gallus-5.0/galGal5). After removing low-quality sequences and adapters, 55.35 Gb of clean reads was obtained with an average GC content of 48.2%. CIRI and circBase were used to identify circRNAs [24-25], and 2,468 circRNAs were identified in regenerating GCs of SYFs in RL and WL groups. Nearly 85% of circRNAs had a predicted splice length of <2,000 bp, 46.64% of circRNAs had a length >500 bp, 26.26% were 500-1000 bp, and the average length was 2,564 bp (Fig. 1A). The identified circRNAs in GCs were distributed across almost all chromosomes, with the fewest on chromosome (chr) 32 and the most on chr1 (Fig. 1B). According to their genomic loci, circRNAs were grouped into exon, intron and intergenic regions. Most (2,180) circRNAs originated from sense genic_exonic regions (Fig. 1C).
Analysis of DE circRNAs
Expression levels of circRNAs of GC libraries of SYFs were compared between RL and WL groups, and 22 DE circRNAs were identified (p <0.05). Of these, 15 were upregulated and 7 were downregulated (Fig. 2A, Additional File 1). Furthermore, spliced reads per million (RPM) was used to measure expression levels of circRNAs [26], and volcano plots of DE circRNAs were plotted (Fig. 2B).
GO and KEGG analyses of host genes related to DE circRNAs
The biological functions of host genes related to DE circRNAs were further analysed. GO enrichment analysis was performed on DE circRNAs involved in the positive regulation of cell migration, the ERK1and ERK2 cascade, cell-cell signalling, and inositol 1,4,5-trisphosphate binding (Fig. 3A, Additional file 2). KEGG pathway enrichment analysis of DE circRNAs identified ovarian steroidogenesis, as well as MAPK signalling, prolactin signalling , PI3K-Akt signalling and thyroid hormone signalling pathways (Fig. 3B, Additional file 3).
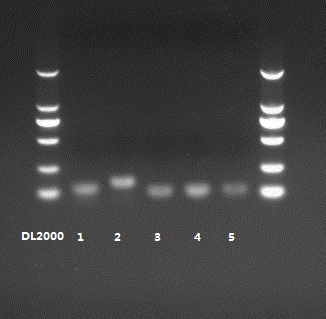
Verification of DE circRNAs and host genes by RT-qPCR
To validate the expression levels of DE circRNAs and host genes to support further analysis of the molecular mechanism of monochromatic light on follicular development in hens, five circRNAs (circRNA_1946, circRNA_0320, circRNA_0185, circRNA_0007, and circRNA_1481) and five genes (FSHR, EGF-like fibronectin type III and laminin G domain [EGFLAM], mitogen-activated protein kinase kinase kinase 5 [MAP3K5], phosphoinositide-3-kinase regulatory subunit 1 [PIK3R1], photopigment melanopsin-like [OPN4-1]) were analysed by RT-qPCR, and the results were compared with the high-throughput RNA-seq results (Fig. 4). The WL group served as a control group, and the expression pattern revealed that the two methods gave consistent results.
Coexpression networks of predicted target miRNAs and genes
It is well established that one of the main functions of circRNAs is acting as miRNA sponges [19], and circRNA-miRNA interactions can predict the characteristics of circRNAs [20]. Herein, 24 miRNAs and 193 mRNAs were predicted by miRBase [52], and the results are shown in Additional files 4 and 5. Cytoscape software was applied to construct circRNA-miRNA-mRNA coexpression networks [53], and circRNA_1805 and circRNA_0007 were found to share the common targets miR-425-5p, circRNA_0320, and circRNA_1805, with the same 11 miRNAs, including miR-143-3p, miR-142-3p, miR-107-3p, miR-27b-3p, and miR-460a-5p. The molecular interactions between these three circRNAs and their miRNAs and target genes are illustrated in Figure 5. Target genes including FSHR, EGFLAM, MAP3K5 and PIK3R1 are related to chicken reproductive performance.
{kind=link}