The detailed approach of selection of IVs for exposures, genome-wide association study (GWAS) summary statistics for ALS, and MR analysis were previously described [15]. The MR approach we used was based on the following three assumptions: 1) genetic variants (single nucleotide polymorphisms (SNPs)) used as IVs are associated with exposures; 2) genetic variants are not associated with confounders; and 3) genetic variants influence the risk of outcomes only through interested exposures , not through other pathways [16] (Figure 1). The IVs (F statistic > 10) for all the exposures were sufficiently informative [17].
Genetically predicted gut microbiota genera
Genetic instruments of the abundance of 98 genera of gut microbiota at the level of genome-wide significance (P < 5×10-8) were obtained from available GWAS data of stool samples in humans [18]. As a result, independently significant SNPs were identified for 22 genera of the gut microbiota, but no significant genetic variants were found for the remaining 76 genera of the gut microbiota.
If an SNP was not available for an outcome, a highly correlated proxy SNP (r2 > 0.9) (https://ldlink.nci.nih.gov/) was used instead, if available. We checked the phenotypes of selected SNPs using comprehensive genotype-to-phenotype cross-references (GWAS Catalog[19]) and repeated the analysis with potentially pleiotropic SNPs excluded. We calculated SNP-specific F statistics as a quotient of squared SNP-genus association and its variance [20].
Genetically predictedgut microbial metabolites
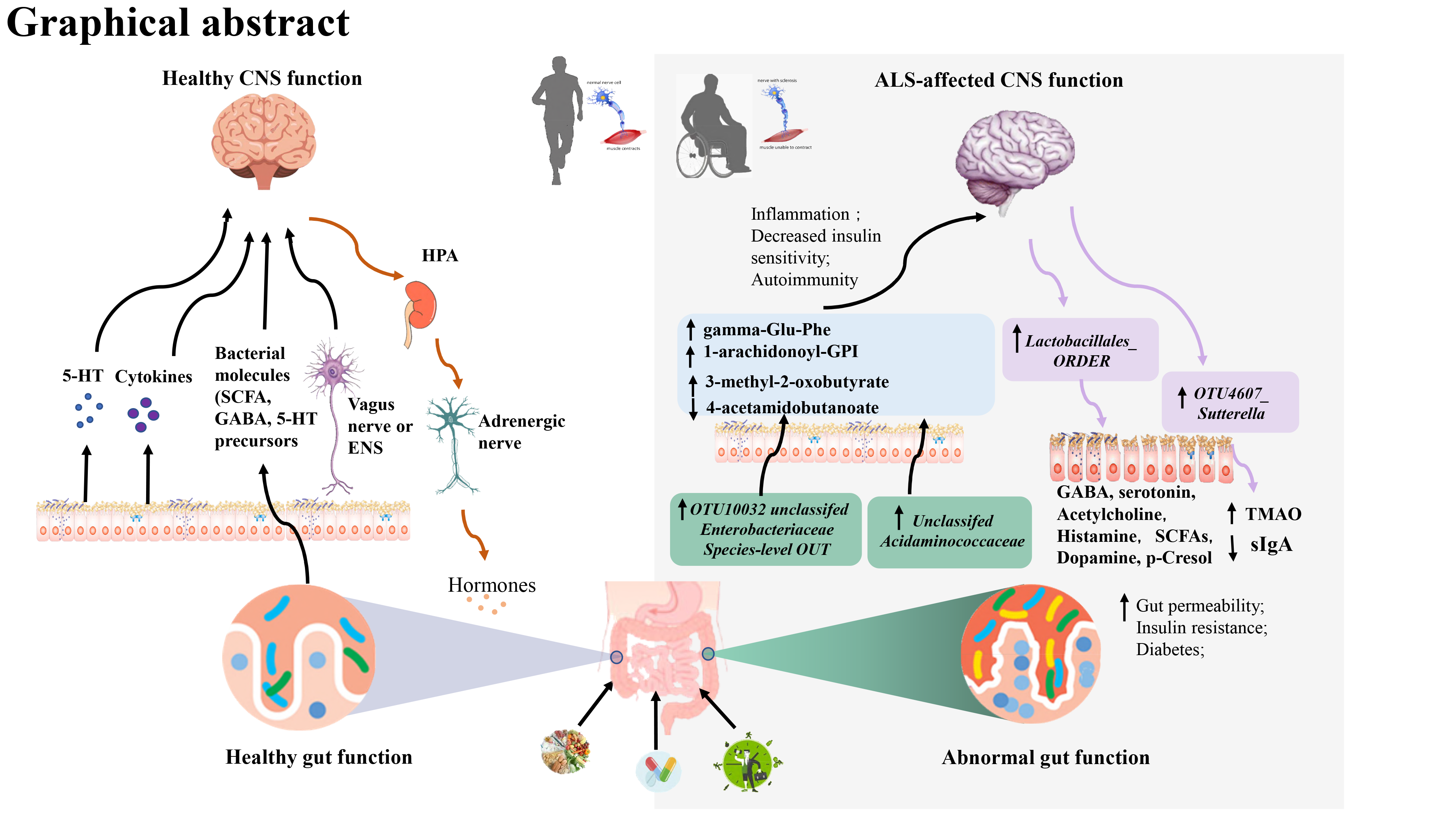
A transsynaptic, glutaminergic, excitotoxic mechanism (the so-called dying-forward hypothesis) has been proposed as a pathophysiological biomarker in ALS [21]. We therefore used 18 potential blood metabolites that might have causal effects on the development of ALS, including a group of gamma-glutamyl amino acids [22]. The candidate metabolites were identified among 486 untargeted serum metabolites from Shin’s study [23]. A total of 7824 adult individuals from 2 European cohorts were included in the GWAS analysis. Metabolomics data were acquired based on nontargeted mass spectrometry analysis of human fasting serum [23].
For each of the metabolites, we selected SNPs that showed an association at P < 1×10-5 as candidate IVs of the specific metabolite. Then, a clumping procedure was conducted with European 1000G as a reference panel to identify the independent variants, with a linkage disequilibrium threshold of r2 < 0.01 in a 500-kb window.
Genetically predicted ALS
We drew on summary statistics from the largest and most recent GWAS of ALS [24] patients who were defined as having been diagnosed with probable or definite ALS according to the El Escorial criteria (Brooks, 1994) by a neurologist specializing in ALS. This GWAS of ALS involving 20,806 patients and 59,804 controls of European ancestry identified 10 independent genome-wide significant SNPs at the level of P < 5×10-8 [24].
Statistical analysis
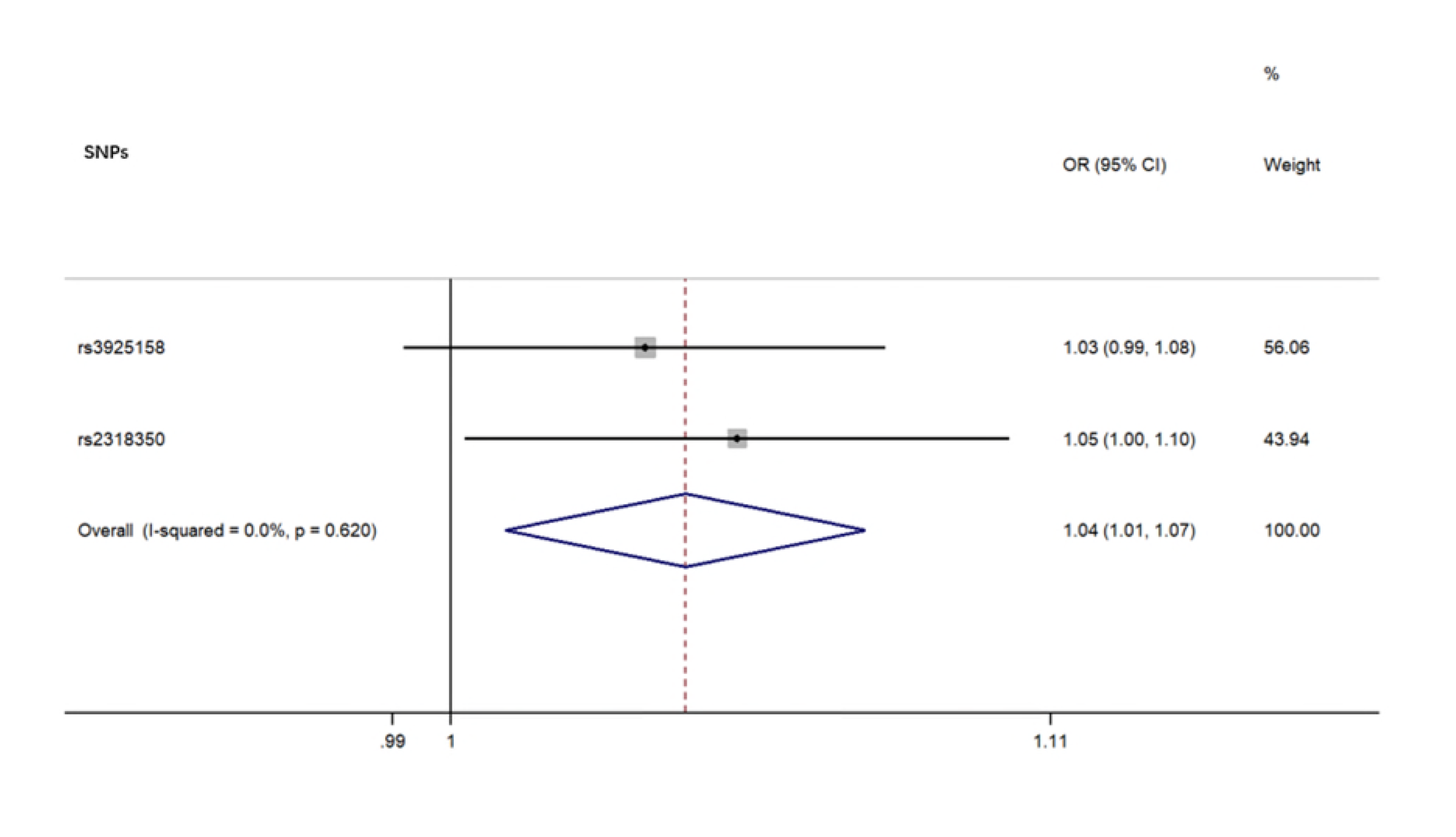
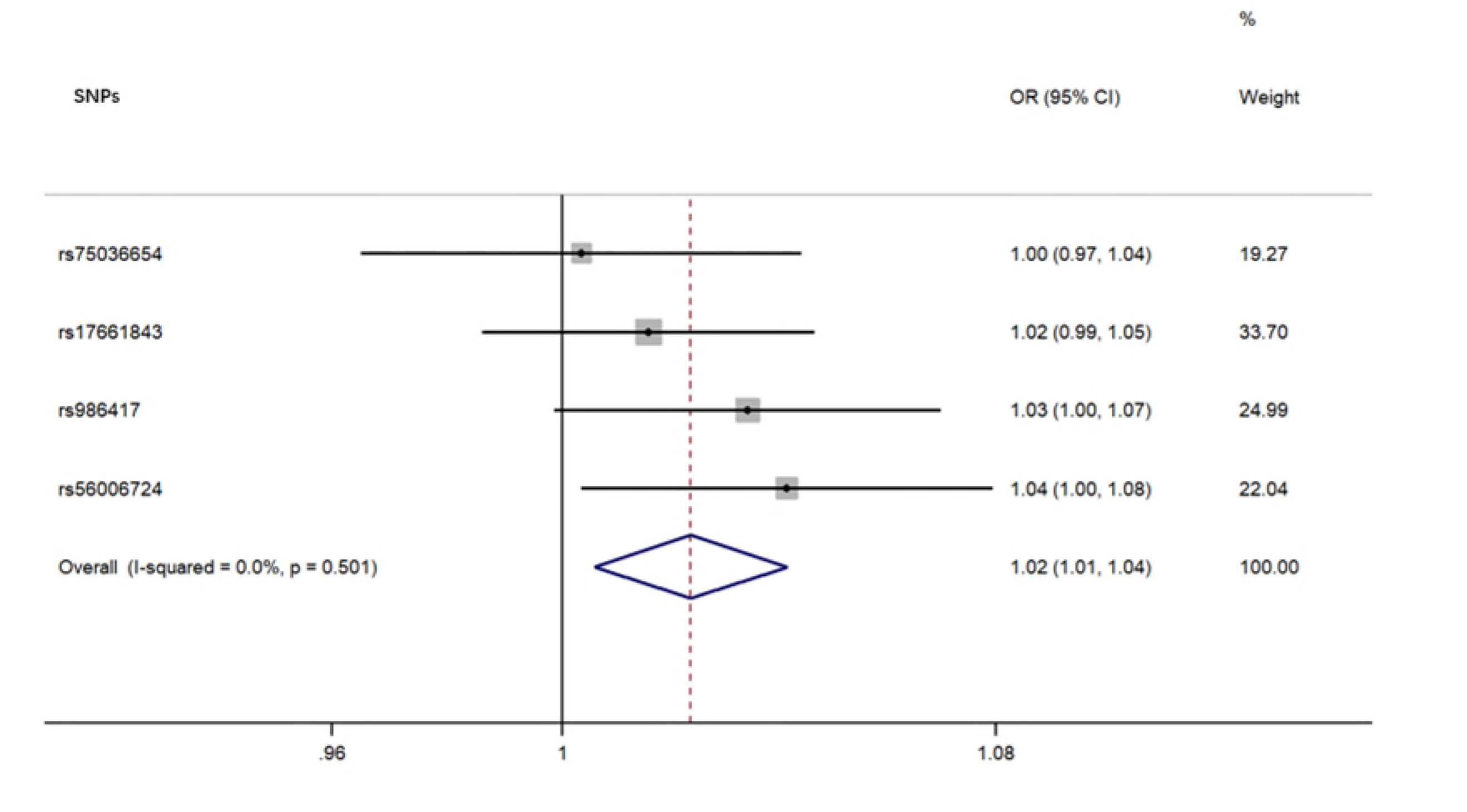
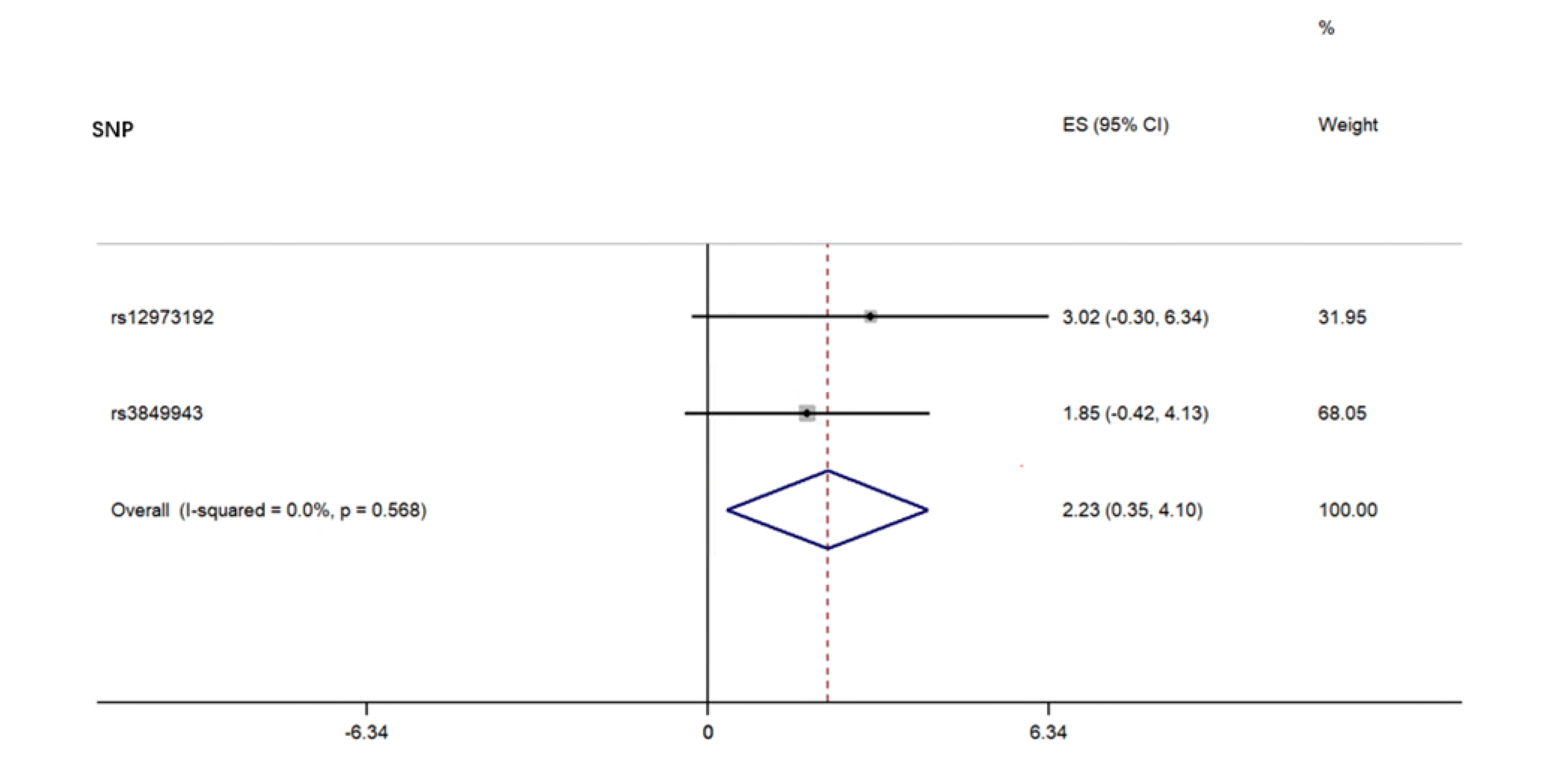
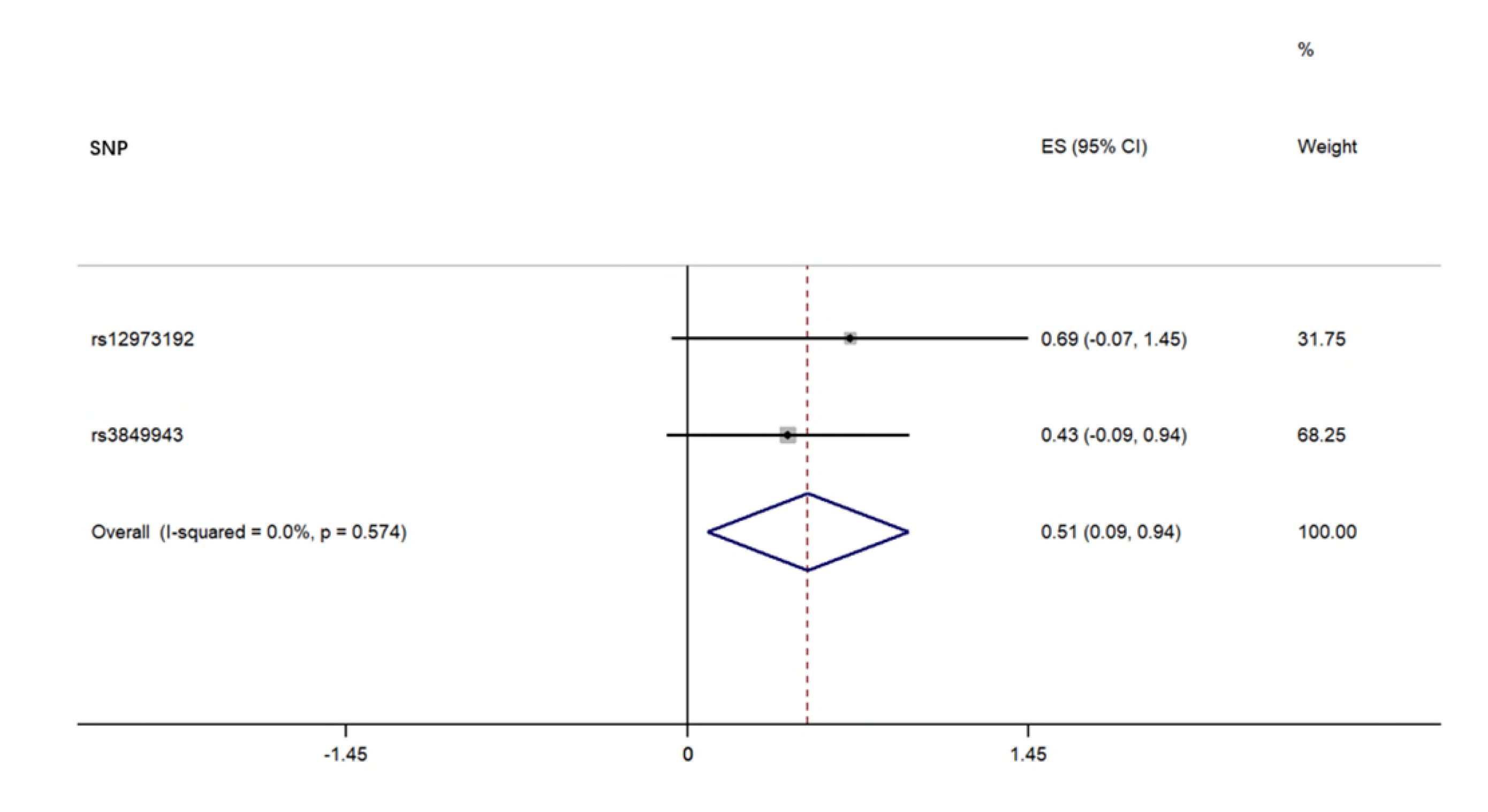
For each direction of the potential relationship, we combined MR estimates using an inverse variance-weighted method (IVW) meta-analysis, which essentially translates to a weighted regression of SNP outcome effects on SNP exposure effects where the intercept is constrained to zero. The IV assumptions can be biased if instrument SNPs show horizontal pleiotropy, influencing the outcome through causal pathways other than exposure [16]. Therefore, other established MR methods, including weighted, weighted median mode, and MR Egger regression, were also applied to confirm the IVW results (number of SNPs ≥3) because their estimates are known to be relatively robust to horizontal pleiotropy, although at the cost of reduced statistical power [25]. MR Egger regression allows the intercept to be freely estimated as an indicator of average pleiotropic bias. Effect estimates are reported in β values when the outcome is continuous (i.e., the abundance of each genus of gut microbiota) and are converted to ORs when the outcome is dichotomous (i.e., ALS status).
To assess the robustness of significant results, we conducted further tests for horizontal pleiotropy using meta-analytic methods to detect heterogeneous outcomes, including leave-1-SNP-out analyses and the MR Egger intercept test of deviation from the null [26].
The analyses were performed with R version 3.1.1 (R foundation) and Stata version 11.2 (Stata Corp, College Station, TX). All human research was approved by the relevant institutional review boards and conducted according to the Declaration of Helsinki. Ethical approval was obtained from relevant Research Ethics Committees and from the review boards of Peking University Third Hospital.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}