Phenotypic variation, Descriptive statistics, and Correlation
The combined analysis of variance revealed significant (p<0.05) genotype and GEI variations for GY, AD, SD, ASI, EPO, EPP, EH, and PH under DS and optimum conditions (Table 1). The mean, minimum, and maximum values of GY, AD, SD, ASI, EPO, EPP, EH, and PH revealed large variability for each trait under both DS and optimum conditions (Table 1). GY under DS was reduced by 71%. PH (214.69 cm) and EH (116.88 cm) were reduced significantly under DS compared to the mean of PH (225.94 cm) and EH (112.27 cm) under optimum conditions. DS had a relatively small effect on EPO and EPP. DS conditions increased the average number of days for AD and SD compared to optimum conditions. Consequently, prolonging the interval between AD and SD (known as ASI) under drought by an average of 3 days compared to optimum conditions. The broad-sense heritability ranged from 0.25 for GY to 0.87 for EPO under DS and varied from 0.21 for EPP to 0.74 for AD under optimum conditions. The frequency distribution of each of the traits under the two experimental conditions (DS and optimum) is shown in Figure 1 where most of the traits showed continuous distributions.
Pearson’s correlation analysis showing the relationships among traits under DS and optimum conditions is presented in Figure 2. The correlation coefficients among the eight traits ranged from -0.29 to 0.93 under optimum conditions, whereas under DS conditions ranged from -0.56 to 0.86. Under optimum conditions, the highest significant positive coefficients correlation (r=0.93**) was observed between flowering traits, SD_OPT and AD_OPT, followed by PH_OPT and EH_OPT (r=0.78), GY_OPT and PH_OPT (r=0.65). GY_OPT had a significant positive correlation with PH_OPT, EH_OPT, and EPP_OPT while a significant but negative correlation was observed between GY_OPT with SD_OPT and ASI_OPT. Similarly, ASI_OPT had a significant negative correlation with AD_OPT, PH_OPT, EH_OPT, and EPP_OPT. Under DS conditions, the highest positive correlations were 0.83 (between GY_DS and EPP_DS), 0.66 (SD_DS and AD_DS), 0.64 (EPO_DS and EH_DS), 0.63 (PH_DS and EH_DS), and 0.57 (EPO_DS and AD_DS). GY_DS was negatively and significantly correlated with AD_DS, SD_DS, ASI_DS, and EPO_DS, while positively and significantly correlated with PH_DS and EPP_DS. It was observed that ASI_DS was positively correlated with AD_DS, SD_DS, and EPO_DS and negatively correlated with PH_DS, EH_DS, and EPP_DS. Additionally, AD_DS was positively correlated with SD_DS, EH_DS and EPO_DS, but negatively correlated with PH_DS and EPP_DS. Overall, a strong correlation was observed between the AD and SD under both optimum and DS conditions.
Marker distribution, Population structure, Phylogenetic tree, and Kinship
The distribution of 215,542 SNPs across the genome is presented in Figure 3A and supplementary Table S2. The number of SNP markers on each chromosome ranged from 14,629 to 33, 874 SNPs. Chromosome 1 had the highest number of SNPs of 33, 874, whereas chromosome 10 had the lowest number of SNPs with 14,629. The density of SNPs per mega-base pair (Mbp) varied from 84.89 Mbp for chromosome 4 to 116.79 Mbp for chromosome 5 with a mean of 104.28 Mbp.
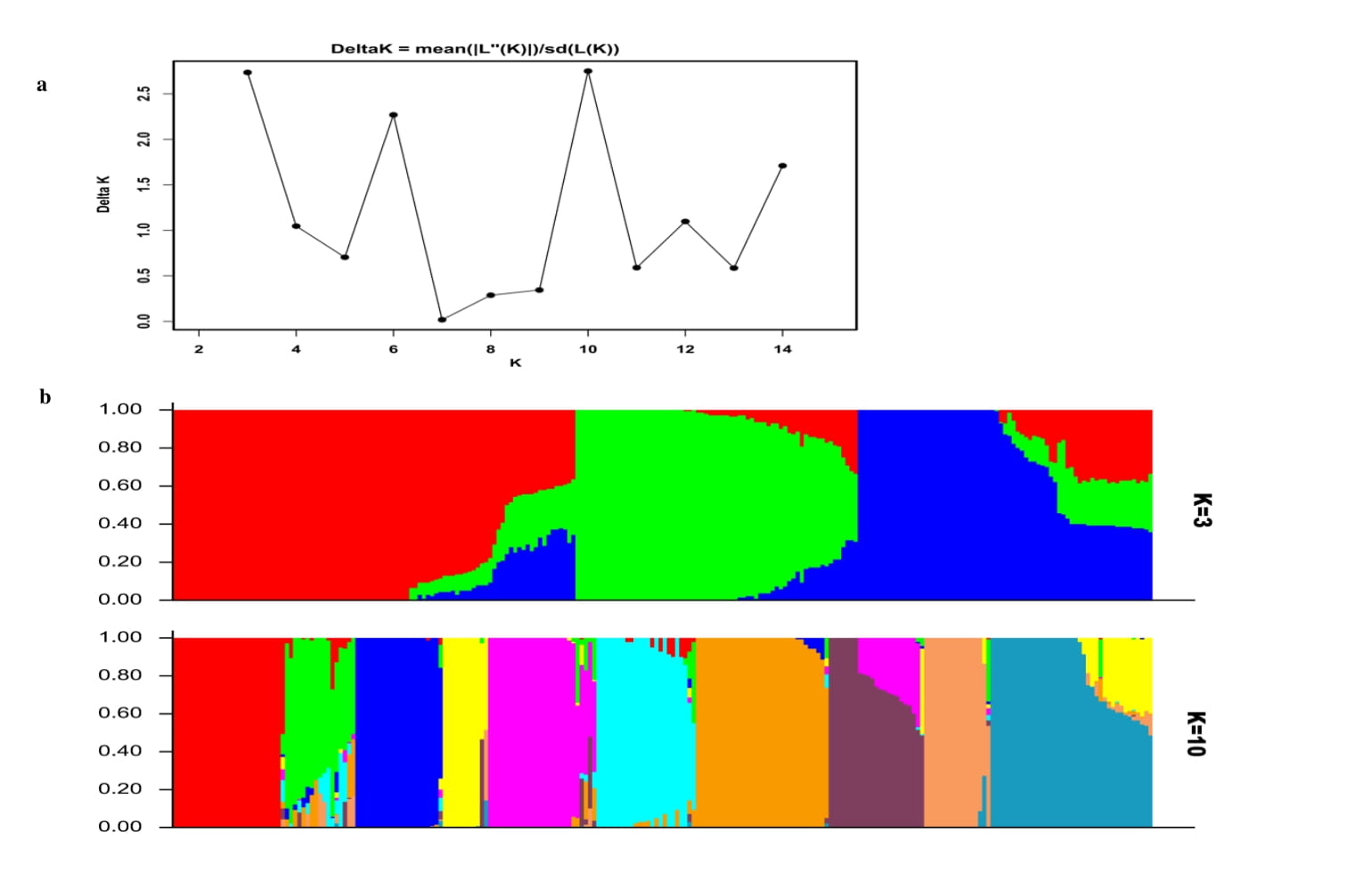
The population structure of 236 diverse maize lines was determined by Bayesian based model in STRUCTURE and PCA (Supplementary Figure S1a and Figure 3B). The optimum number of K was obtained by plotting the number of clusters (K) against delta K (Supplementary Figure S1a). The Bayesian structure analysis revealed the presence of three distinct subgroups within the 236 maize lines when K = 3 and ten distinct subgroups K = 10 (Supplementary Figure S1b). The genetic structure was further examined by PCA (Figure 3B). The results revealed the presence of four distinct subpopulations separated by PC1 (12.78 %), PC2 (9.11%), and PC3 (7.19 %). The first three PCs explained over 29 % of the total genetic variation among the diverse maize lines. A scree plot of variance explained against the corresponding PCs as shown in Figure 3C was used to determine the optimal number of PCs to retain. The scree plot revealed that an optimal number (K) of four PCs could be retained for GWAS. The phylogenetic tree based on the neighbor-joining method as shown in Figure 3d revealed that the 236 diverse maize lines can be clustered into four main groups (I=66, II=48, III=100, and IV=22) differentiated by the different colors (Supplementary Table S1).
The kinship matrix is utilized to assess the relatedness among individuals by considering the extent of allele sharing. The pattern of red shading in the center of the kinship matrix (Figure 3F reflected the level of genetic relatedness among individuals, indicating the presence of four stratified population structures. In general, the results from the PCA, phylogenetic tree, and kinship matrix, show that the panel of 236 diverse maize lines can be divided into four subpopulations.
Linkage Disequilibrium and GWAS analysis
The nonrandom association of alleles between two genetic loci was examined to provide valuable information in locating genes tightly linked to SNP markers associated with traits of interest. A rapid LD decay pattern was observed across the ten chromosomes (Figure 3F). At r2 = 0.2, the LD decay distance between SNPs on the 10 chromosomes varied from 2.83 kb (Chr3) to 11.83 kb (Chr4) with an average genome-wide LD decay with physical distance between SNPs of 4.74 kb.
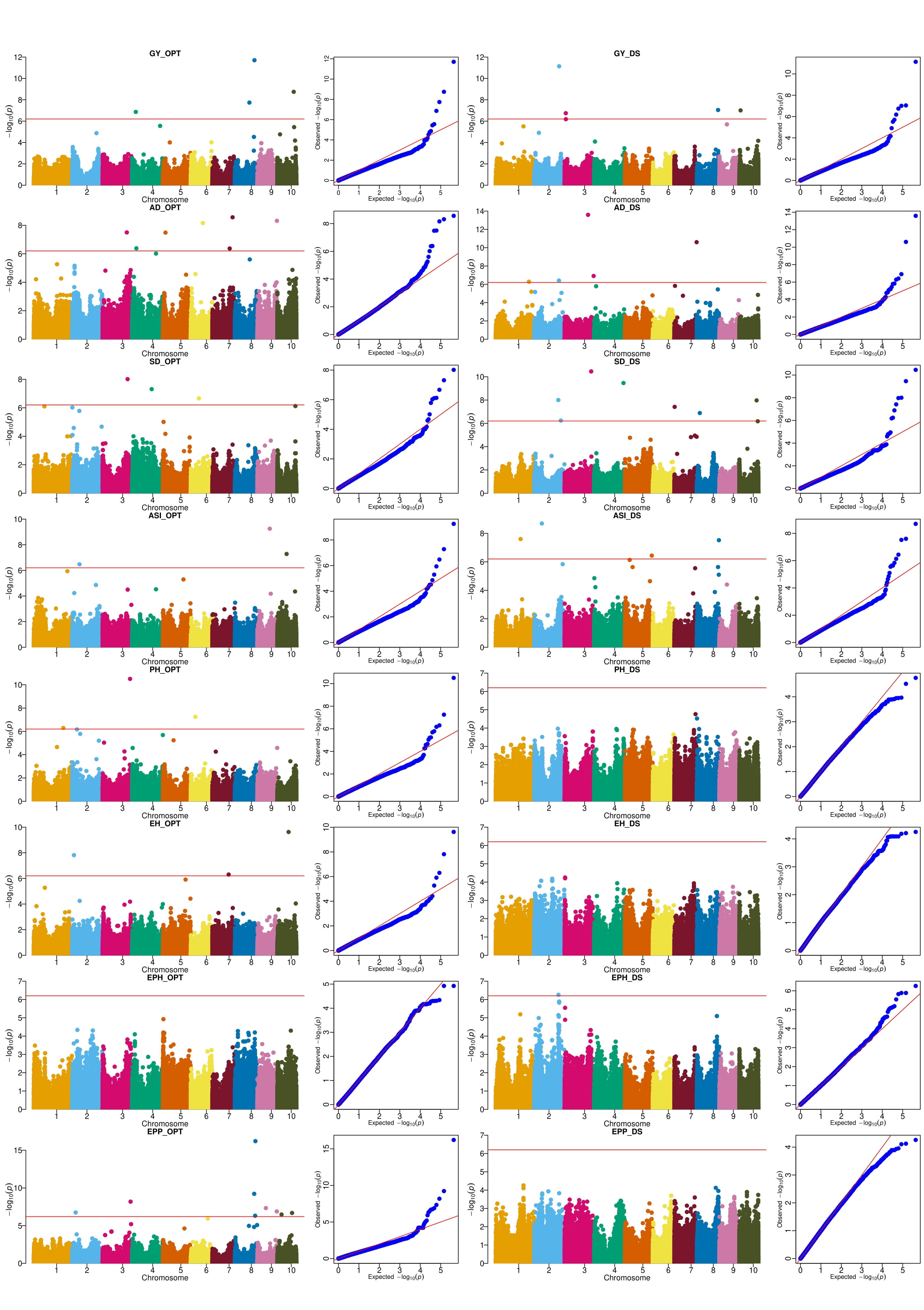
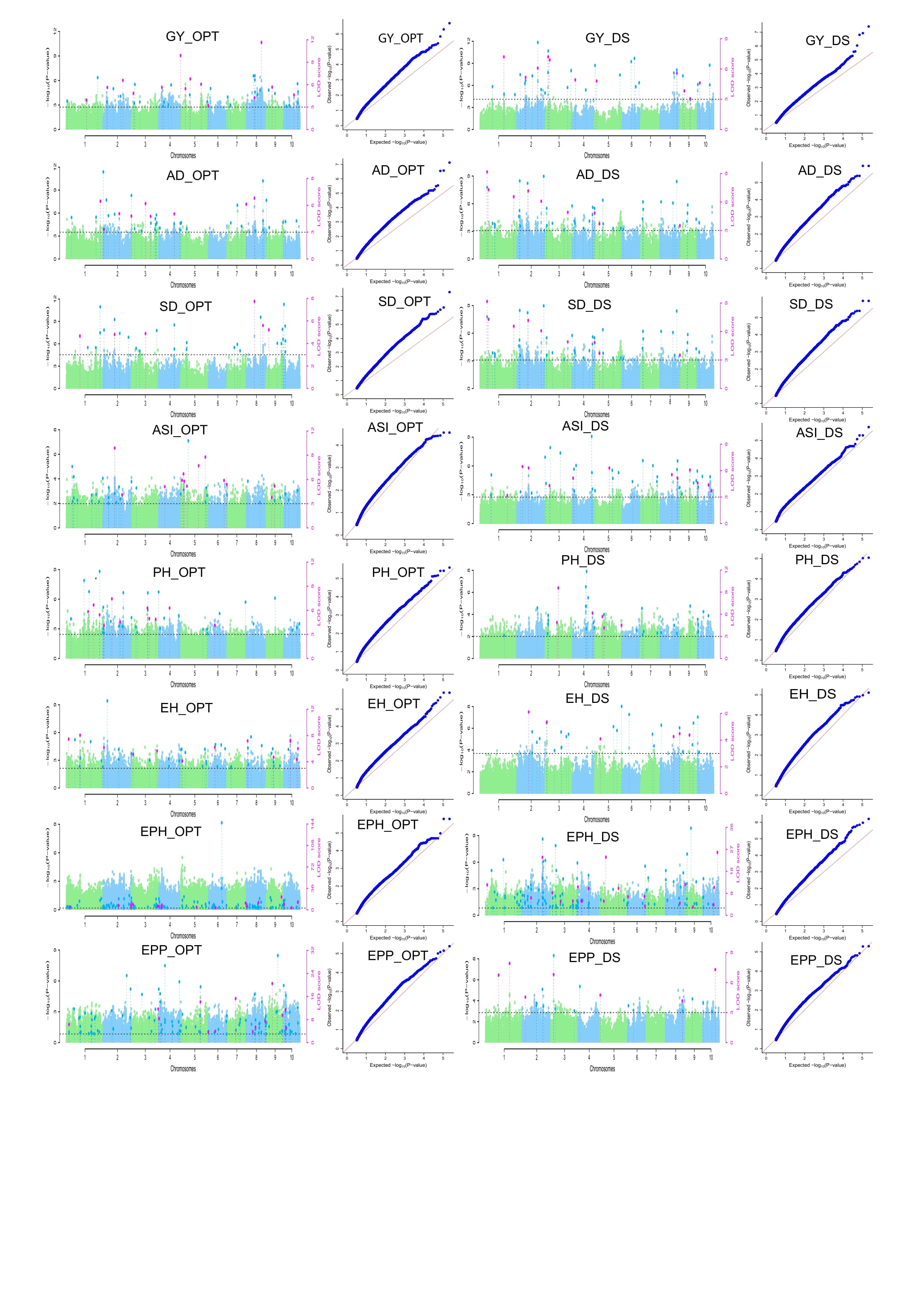
Based on the seven multi-locus GWAS models (FarmCPU, mrMLM, FastmrMLM, FASTmrEMMA, pLARmEB, pKWmEB and ISIS EM-BLASSO), 219 and 265 QTNs distributed across the 10 chromosomes were detected under DS and optimum conditions, respectively (Figure 4, Supplementary Table S3 and S4). The QQ-plots depicting the observed vs expected LOD [−log10 (p), p= 6.29e-07] distributions showed a significant deviation from the expected distribution (Supplementary Figure S2 and S3), indicating the presence of QTNs linked to GY, AD, SD, ASI, PH, EH, EPP and EPO, respectively under DS and optimum conditions. The Manhattan plot showed that several QTNs with Bonferroni-adjusted threshold of greater than 6.20 [−log10 (p), p= 6.29e-07] and LOD threshold of ≥ 3 (p = 0.0002), were associated with GY, AD, SD, ASI, PH, EH, EPP and EPO, respectively under DS and optimum conditions.
The number of significant QTNs detected by multiple GWAS models varied across traits. Of these, the power of detection among the seven models followed FASTmrMLM (62 QTNs) > pLARmEB (45 QTNs) > ISIS EM-BLASSO (37 QTNs) > pKWmEB (36 QTNs) > FarmCPU (16 QTNs) > FASTmrEMMA (12 QTNs) > mrMLM (7 QTNs) under DS and FASTmrMLM (68 QTNs) > pLARmEB (62 QTNs) > pKWmEB (42 QTNs) > FASTmrEMMA (29 QTNs) > FarmCPU (25 QTNs) >ISIS EM-BLASSO (24 QTNs) > mrMLM (15 QTNs) under optimum conditions (Fig. 4C and 4D). Of these QTNs, 46, 33, 31, 30, 29, 20, 17, and 13 were found to be associated with EPO, ASI, AD, GY, SD, EPP, EH and PH under DS, respectively. Under optimum conditions, 40, 37, 36, 36, 32, 25, and 24 QTNs were associated with EPO, EH, ASI, GY, EPP, PH, SD and AD, respectively (Fig. 4A and 4B). With the proportion of R2 (%) by individual QTNs accounting for 0.00 ~ 8.18, 0.60 ~ 16.06, 3.93 ~ 15.29, 1.43 ~30.68, 0.00 ~ 11.42, 0.75 ~ 10.08, 1.32 ~ 16.06 and 2.00 ~ 12.66 (%) of total phenotypic variation for EPO, EH, ASI, GY, EPP, PH, SD and AD, respectively (Supplementary Table S4, S5 and S6).
Significant QTNs detected by at least two ML-GWAS models were considered reliable and stable. A sum of 172 QTNs comprising 77 QTNs under DS and 95 QTNs under optimum conditions were discovered to be significantly associated with EPO, ASI, AD, GY, SD, EPP, EH, and PH, respectively (Fig. 4E and 4F; Supplementary Table S6). Of these significant QTNs under DS, 9, 9, 9, 11, 6, 6, 7, and 20 reliable QTNs were detected for GY, AD, SD, ASI, PH, EH, EPP, and EPO, respectively. The highest R2 (%) value was observed for GY QTN qGY_DS2.1 [S2_194907656:R2 =5.07 %-11.07%], for AD qAD_DS1.2 [S1_32228028: R2 =5.27%-11.27%], for SD qSD_DS10.1 [S10_2032146:R2 = 5.36 % - 9.92 %], for ASI qASI_DS5.1 [S5_151886720:R2 =5.94 %-11.19 %], for PH qPH_DS3.2 [S3_149683959:R2 = 9.62%-11.27 %], for EH qEH_DS2.1 [S2_60655022:R2 = 6.23%-6.78%], for EPP qEPP_DS1.1 [S1_217115118:R2 = 9.19 %-13.26 %] and for EPO qEPH_DS2.1 [S2_36458273:R2 = 0.96 % - 6.15 %].
In addition, of the 95 QTNs detected in optimum condition, 12, 9, 6, 13, 13, 11, 18, and 13 QTNs were found to be associated with GY, AD, SD, ASI, EH, PH, EPO, and EPP, respectively. The QTN, qGY_OPT5.2 [S5_193538664: R2 = 5.34% - 10.19%] recorded the highest R2 (%) for GY. One QTN, qAD_OPT8.1 (S8_105322358) was shared by AD (R2 = 6.03 % -12.66 %) and SD (R2 = 3.99 %-16.06 %). For ASI, qASI_OPT6.1 [S6_159006932: R2 = 6.30% - 14.10%] explained the highest R2 (%). While qEH_OPT10.4 [S10_88760138: R2 = 9.39 % - 16.06 %] showed the highest R2 (%) for EH, qPH_OPT3.1 [S3_172201680: R2 = 1.49 % - 10.08%] for PH, qEPO_OPT2.2 [S2_233748945: R2 = 1.56 % - 3.26 %] for EPO and qEPP_OPT8.2 [S8_75600223: R2 = 0.002 % -5.48 %] for EPP (Figure 4 and Figure 5).
The detected QTNs contributing revealed positive and negative allelic effects for different traits (Figure 4E and Figure 5). Among the 172 QTNs, few QTNs were detected under both DS and optimum conditions. For instance, two QTNs qGY_DS2.1 and qGY_DS1.1 explained the highest and positive effects with 0.34 and 0.24, respectively under DS for GY. On the other hand, two new QTNs qGY_OPT4.1 and qGY_OPT10.1 showed the highest and positive effects of 0.56 and 0.49, respectively for GY under optimum conditions. Negative QTN effects are desirable for flowering traits (AD, SD, and ASI) under DS for selecting earliness. For instance, QTNs qAD_DS2.1, qSD_DS8.1, qASI_DS1.1 exhibited large and negative effects for AD (-1.72), SD (-1.79), and ASI (-0.77) on chromosome 2, 8 and 1, respectively (Figure 4E). The distribution of QTNs (Figure 5) on the 10 chromosomes showed that chromosome 5 captured the higher number of reliable QTNs (12 under DS and 14 under optimum conditions). Two QTNs, qPH_OPT2.2 and qEH_OPT2.1 exhibited a pleiotropic effect on both PH and EH under optimum conditions.
Candidate gene identification and Annotation
Candidate genes analysis was conducted for significant QTNs identified in this study to elucidate the molecular, biological, and physiological mechanisms controlling traits under DS and optimum conditions. A total of 43 candidate genes were discovered and annotated, among them 18 and 25 candidate genes were identified under DS and optimum conditions, respectively (Table 2). Two candidate genes closely associated with the QTNs for improved GY were identified under DS, namely, Zm00001eb041070 and Zm00001d045665 (Table 2). Nine candidate genes potentially associated with flowering traits (AD, SD and ASI) were identified under DS (Table 2). Of these five candidate genes were associated with ASI under DS. Similarly, four and three candidate genes were found to be associated with EPO and EPP, respectively. Under optimum conditions, 3, 5, 8, 3, and 3 candidate genes were identified for GY, PH, EH, EPO, and EPP, respectively (Table 2). On the other hand, one candidate gene each was identified for AD, SD, and ASI.
Haplotype Analysis
The QTN qGY_DS1.1 (S1_216149215) associated with GY under DS was identified in a 216.15 Mb region on chromosome 1 based on pairwise LD correlation (Figure 6A and 6C). The QTN region contains six distinct SNPs with relatively high LD (Figure 6C). Four major haplotypes were detected among the 236 lines with Haplotypes frequencies of 142 (60 %), 39 (17 %), 29 (12 %), and 7 (3%) for Hap1 (GAGGGC), Hap2 (AAGGGC), Hap3 (GTAATG), and Hap4 (GAGGTG), respectively (Figure 6B). A significant difference was observed between haplotypes Hap1 and Hap2 (Figure 6D). The mean GY was 2.2 t/ha for Hap1, 1.80 t/ha for Hap2, 2.10 t/ha for Hap3, and 2.2 t/ha for Hap4, respectively. (Figure 6D). Hap1 was considered a superior haplotype since it contributes to the highest mean performance compared to other haplotypes.
Genomic prediction
Among the GP models, RR-BLUP is computationally less intensive and is well suited for routine application in plant breeding trials. Therefore, we used the RR-BLUP model to estimate the performance of maize genotypes for various traits under optimum and DS conditions (Figure 7). Prediction accuracies were moderate to high for all eight traits under optimum and low to moderate under DS conditions (Figure 7). The observed prediction accuracy for GY, AD, SD, ASI, PH, EH, EPO, and EPP were 0.29, 0.58, 0.45, 0.44, 0.61, 0.54, 0.24, and 0.25, respectively under DS conditions, and 0.65, 0.72, 0.69, 0.37, 0.71, 0.51, 0.48, and 0.41, under optimum conditions.
{kind=link}
{kind=link}
{kind=link}