Materials
All chemicals and reagents used for synthesis were purchased from Loba Chemie, Sigma Aldrich, and SD Fine Chemicals Pvt. Ltd, India. All the reactions were monitored using thin layer chromatography (TLC) on pre-coated TLC plates (Silica gel GF254) using various solvent systems. Melting points were taken in open capillary tubes using ANALAB μThermoCal10 melting point apparatus and were uncorrected. The structures of the synthesized compounds were confirmed by Infrared (IR) spectroscopy and Nuclear Magnetic Resonance (NMR) spectroscopy. IR spectroscopy was carried out using KBr pellet method on the Perkin Elmer Spectrum 10.4.2 and Shimadzu IRAffinity-1. NMR spectra were recorded on a Bruker Avance 500 spectrophotometer operating at 500 MHz (1H and 13C NMR). Samples were dissolved (20mg/ml) in hexadeuterated dimethylsulfoxide (DMSO-d6), and spectra were recorded at 323 K. Column chromatography was carried out using silica gel (230/400 mesh).
Methods
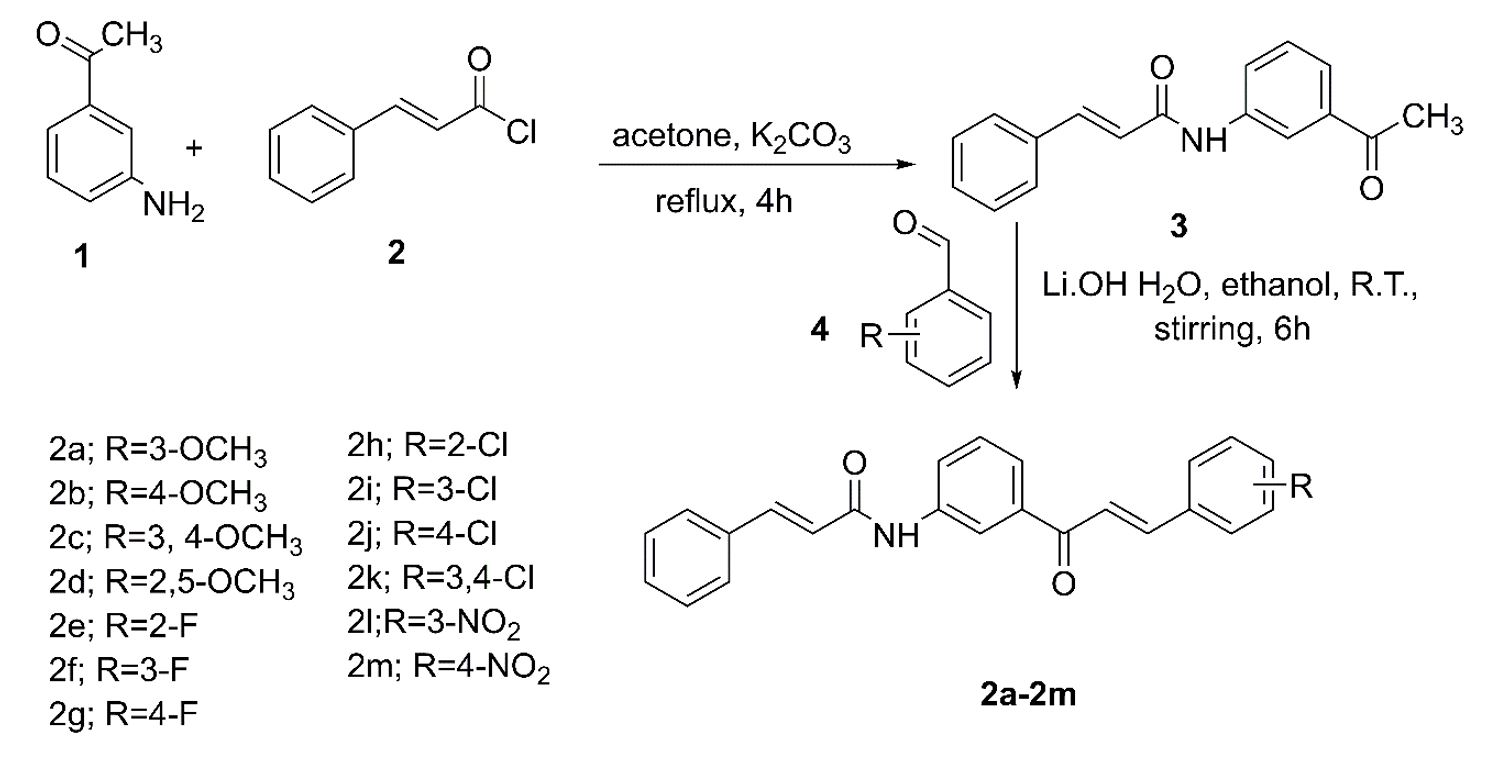
Synthesis of cinnamamide chalcones (2a-2m)
A mixture of 3-aminoacetophenone (0.01moles) and cinnamoyl chloride (0.01moles) was refluxed under acetone (20 ml) and potassium carbonate (0.001moles) for 4 h. The completion of the reaction was monitored by TLC. The mixture was then decanted to remove potassium carbonate and then allowed to stand overnight to obtain a white solid of N-(3-acetylphenyl) cinnamamide [40, 41]. It was then filtered under vacuum to obtain the dry intermediate of N-(3-acetylphenyl) cinnamamide.
N-(3-acetylphenyl) cinnamamide in anhydrous ethanol (20 ml) was treated with lithium hydroxide monohydrate (0.003 moles) under magnetically stirred condition for 15 min at room temperature. The substituted benzaldehyde (0.01moles) was added to the stirring mixture [42]. Stirring was continued for 6 h. The reaction was confirmed for completion by TLC. The reaction mixture was acidified with 1% aqueous HCl to give crude product. The resulting product was filtered and recrystallized from ethanol.
1-(3’-cinnamaamidophenyl)-3-(3”-methoxyphenyl)-prop-2-en-1-one (2a)
White (pale) colored solid, yield (69.20%). mp:169°C; FTIR (KBr, cm-1): 3344.57 (N-H stretch of anilide), 1662.64 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 1251.8 (asymmetric C-O-C stretch, OCH3), 1033.85 (symmetric C-O-C stretch, OCH3); 1H NMR (DMSO-d6, δ ppm): 10.299 (s, 1H, NH), 8.188 (s, 1H, H-2’), 7.904 (d, 1H, H-4’), 7.788 (d, 1H, H-6’), 7.734 (d, 1H, H-3), 7.598 (d, 1H, H-2), 7.503 (m, 3H, H-2’’’,6’’’,6), 7.427 (t, 1H, H-5’), 7.323 (m, 5H, H-3’’’,4’’’,5’’’,6’’,2’’), 7.252 (t, 1H, H-5’’), 6.905 (d, 1H, H-4’’), 6.716 (d, 1H, H-5), 3.686 (s, 3H, OCH3); 13C NMR δ ppm 189.580 (C-1), 164.199 (C-4), 160.064 (C-3’’), 144.526 (C-3), 140.978 (C-6), 140.145 (C-1’), 138.549(C-3’), 136.429 (C-1’’’), 135.014 (C-1’’), 130.348 (C-5’), 130.262 (C-5’’), 129.712 (C-4’’’,4’), 129.416 (C-3’’’,5’’’), 128.161 (C-2’’’,6’’’), 124.163 (C-6’), 122.863 (C-2), 122.377 (C-6’’), 121.881 (C-2’), 119.197 (C-5), 117.067 (C-4’’), 113.908 (C-2’’), 55.697 (-OCH3).
1-(3’-cinnamaamidophenyl)-3-(4”-methoxyphenyl)-prop-2-en-1-one (2b)
Yellow colored solid, yield (76.12%). mp:176°C; FTIR (KBr, cm-1): 3322.99 (N-H stretch of anilide), 1656.85 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 1251.8 (asymmetric C-O-C stretch, OCH3), 1033.85 (symmetric C-O-C stretch, OCH3); 1H NMR (DMSO-d6, δ ppm): 10.286 (s, 1H, NH), 8.175 (s, 1H, H-2’), 7.883 (d, 1H, H-4’), 7.749 (d, 1H, H-6’), 7.704 (d, 2H, H-2’’’,6’’’), 7.582 (d, 1H, H-3), 7.575 (d, 1H, H-2), 7.501 (m, 3H, H-6,2’’,6’’), 7.410 (t, 1H, H-5’), 7.329 (m, 3H, H-3’’’,4’’’,5’’’), 6.895(d, 1H, H-3’’,5’’), 6.713 (d, 1H, H-5), 3.682 (s, 3H, OCH3); 13C NMR δ ppm 189.362 (C-1), 164.187 (C-4), 161.832 (C-4’’), 144.558 (C-3), 140.956 (C-6), 140.095 (C-1’), 138.864(C-3’), 135.019 (C-1’’’), 131.121 (C-2’’,6’’), 130.268 (C-5’), 129.669 (C-4’’’), 129.424 (C-3’’’,5’’’), 128.162 (C-2’’’,6’’’), 127.642 (C-4’,1’’), 123.970 (C-6’), 122.397 (C-2), 120.009 (C-2’), 119.130 (C-5), 114.859 (C-3’’,5’’), 55.778 (OCH3).
1-(3’-cinnamaamidophenyl)-3-(3’’,4”-dimethoxyphenyl)-prop-2-en-1-one (2c)
Yellow (bright) colored solid, yield (58.52%). mp:180°C; FTIR (KBr, cm-1): 3319.49 (N-H stretch of anilide), 1656.85 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 1251.8 (asymmetric C-O-C stretch, OCH3), 1033.85 (symmetric C-O-C stretch, OCH3); 1H NMR (DMSO-d6, δ ppm): 10.290 (s, 1H, NH), 8.149 (s, 1H, H-2’), 7.901 (d, 1H, H-4’), 7.775 (d, 1H, H-6’), 7.622 (d, 1H, H-3), 7.581 (d, 1H, H-2), 7.501 (m, 3H, H-2’’’,6’’’,6), 7.420 (m, 2H, H-5’,2’’), 7.329 (m, 4H, H-3’’’,4’’’,5’’’,6’’), 6.899(d, 1H, H-5’’), 6.718 (d, 1H, H-5), 3.714 and 3.677 (s, 6H, OCH3); 13C NMR δ ppm 189.458 (C-1), 164.183 (C-4), 151.752 (C-3’’), 149.437 (C-4’’), 145.109 (C-3), 140.946 (C-6), 140.094 (C-1’), 138.933 (C-3’), 135.020 (C-1’’’), 130.257 (C-5’), 129.617 (C-4’’’), 129.418 (C-3’’’,5’’’), 128.158 (C-2’’’,6’’’), 127.858 (C-4’), 124.196 (C-1’’), 124.070 (C-6’), 123.930 (C-6’’), 122.407 (C-2), 120.179 (C-2’), 119.104 (C-5), 112.015 (C-5’’), 111.312 (C-2’’), 56.143 (3’’-OCH3), 55.998 (4’’-OCH3).
1-(3’-cinnamaamidophenyl)-3-(2’’,5”-dimethoxyphenyl)-prop-2-en-1-one (2d)
Yellow colored solid, yield (71.19%). mp:175°C; FTIR (KBr, cm-1): 3319.42 (N-H stretch of anilide), 1662.44 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 1251.8 (asymmetric C-O-C stretch, OCH3), 1031.92 (symmetric C-O-C stretch, OCH3); 1H NMR (DMSO-d6, δ ppm): 10.473 (s, 1H, NH), 8.345 (s, 1H, H-2’), 8.053 (m, 2H, H-4’,3), 7.931 (m, 2H, H-6’,2), 7.666 (m, 3H, H-2’’’,6’’’,6), 7.583 (m, 2H, H-5’,3’’), 7.474 (m, 4H, H-3’’’,4’’’,5’’’,4’’), 7.071 (s, 1H, H-6’’), 6.879 (d, 1H, H-6), 3.870 and 3.811 (s, 6H, OCH3); 13C NMR δ ppm 189.721 (C-1), 164.300 (C-4), 153.719 (C-5’’), 153.290 (C-2’’), 141.091 (C-6), 140.218 (C-3), 139.076 (C-1’), 139.038 (C-3’), 137.087 (C-1’’’), 135.089 (C-5’), 130.400 (C-4’’’), 130.330 (C-4’), 129.526 (C-3’’’,5’’’), 128.290 (C-2’’’,6’’’), 124.144 (C-6’,2), 122.740 (C-2’), 122.467 (C-5), 119.212 (C-1’’), 118.589 (C-3’’), 113.412 (C-4’’,6’’), 56.629 (2’’-OCH3), 56.185 (5’’-OCH3).
1-(3’-cinnamaamidophenyl)-3-(2’’-fluorophenyl)-prop-2-en-1-one (2e)
White (pale) colored solid, yield (89.92%). mp:158°C; FTIR (KBr, cm-1): 3341.41 (N-H stretch of anilide), 1659.96 (C=O stretch of anilide), 1599.77 (C=O stretch of α, β unsaturated ketone), 1516.31 (aromatic C=C stretch), 752.0 (C-F stretch) ; 1H NMR (DMSO-d6, δ ppm): 10.491 (s, 1H, NH), 8.383 (s, 1H, H-2’), 8.115 (t, 1H, H-5’), 8.059 (d, 1H, H-4’), 7.950 (m, 2H, H-6’,3), 7.866 (d, 1H, H-2), 7.666 (m, 3H, H-2’’’,6’’’,6), 7.596 (m, 2H, H-3’’,5’’), 7.489 (m, 3H, H-3’’’,4’’’,5’’’), 7.379 (m, 2H, H-4’’,6’’), 6.875 (d, 1H, H-5); 13C NMR δ ppm 189.356 (C-1), 164.235 (C-4), 162.368 (C-2’’), 141.026 (C-3), 140.225 (C-6), 138.288 (C-1’), 135.938 (C-3’), 135.010 (C-1’’’), 133.065 (C-5’), 130.286 (C-4’’), 129.820 (C-4’’’,6’’), 129.430 (C-3’’’,5’’’), 128.179 (C-2’’’,6’’’), 125.418 (C-4’), 124.727 (C-5’’), 124.301 (C-6’), 124.103 (C-1’’), 122.612 (C-2), 122.351 (C-2’), 119.195 (C-5), 116.456 (C-3’’).
1-(3’-cinnamaamidophenyl)-3-(3’’-fluorophenyl)-prop-2-en-1-one (2f)
Yellow colored solid, yield (93.52%). mp:155°C; FTIR (KBr, cm-1): 3340.15 (N-H stretch of anilide), 1662.49 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch); 1H NMR (DMSO-d6, δ ppm): 10.477 (s, 1H, NH), 8.376 (s, 1H, H-2’), 8.078 (d, 1H, H-4’), 7.971 (m, 2H, H-2’’’,3), 7.856 (d, 1H, H-6’), 7.784 (d, 1H, H-2), 7.713 (d, 1H, H-6’’’), 7.674 (m, 3H, H-6,6’’,2’’), 7.588 (t, 1H, H-5’), 7.535 (m, 4H, H-3’’’,4’’’,5’’’,4’’), 7.320 (t, 1H, H-5’’), 6.884 (d, 1H, H-5); 13C NMR δ ppm 189.427 (C-1), 164.214 (C-4), 161.920 (C-3’’), 142.989 (C-3), 140.988 (C-6), 140.184 (C-1’), 138.370 (C-1’’), 137.633 (C-3’), 135.021 (C-1’’’), 131.243 (C-5’), 130.228 (C-5’’), 129.701 (C-4’’’), 129.386 (C-3’’’,5’’’), 128.153 (C-2’’’,6’’’), 125.784 (C-4’), 124.309 (C-6’), 123.994 (C-6’’), 122.382 (C-2), 119.256 (C-2’), 117.738 (C-5), 115.152 (C-4’’), 114.977 (C-2’’).
1-(3’-cinnamaamidophenyl)-3-(4’’-fluorophenyl)-prop-2-en-1-one (2g)
Yellow (very light) colored solid, yield (79.13%). mp:168°C; FTIR (KBr, cm-1): 3344.52 (N-H stretch of anilide), 3001.17 (aromatic C-H stretch), 1658.78 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch); 1H NMR (DMSO-d6, δ ppm): 10.469 (s, 1H, NH), 8.359 (s, 1H, H-2’), 8.057 (d, 1H, H-4’), 7.995 (m, 3H, H-2’’’,6’’’,6’), 7.867 (d, 1H, H-3), 7.793 (d, 1H, H-2), 7.664 (m, 3H, H-6,2’’,6’’), 7.583 (t, 1H, H-5’), 7.485 (m, 3H, H-3’’’,4’’’,5’’’), 7.346 (m, 2H, H-3’’,5’’), 6.876 (d, 1H, H-5); 13C NMR δ ppm 189.421 (C-1), 164.198 (C-4), 162.835 (C-4’’), 143.275 (C-3), 140.969 (C-6), 140.153 (C-1’), 138.533 (C-3’), 135.013 (C-1’’’), 131.696 (C-5’), 131.579 (C-2’’,6’’), 131.516 (C-1’’), 130.241 (C-4’’’), 129.692 (C-4’), 129.398 (C-3’’’,5’’’), 128.153 (C-2’’’,6’’’), 124.137 (C-6’), 122.385 (C-2’,2), 119.197 (C-5), 116.430 (C-5’’), 116.257 (C-3’’).
1-(3’-cinnamaamidophenyl)-3-(2’’-chlorophenyl)-prop-2-en-1-one (2h)
Buff colored solid, yield (89.65%). mp:167°C; FTIR (KBr, cm-1): 3344.52 (N-H stretch of anilide), 3001.17 (aromatic C-H stretch), 1658.78 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch); 1H NMR (DMSO-d6, δ ppm): 10.308 (s, 1H, NH), 8.215 (s, 1H, H-2’), 8.043 (d, 1H, H-4’), 7.915 (m, 2H, H-2’’’,3), 7.799 (m, 2H, H-6’’’,2), 7.504 (m, 2H, H-5’,6’), 7.446 (m, 3H, H-6, 3’’,5’’), 7.363 (m, 5H, H-3’’’,4’’’,5’’’,4’’,6’’), 6.717 (d, 1H, H-5); 13C NMR δ ppm 189.291 (C-1), 164.215 (C-4), 141.016 (C-3), 140.215 (C-6), 139.049 (C-1’), 138.228 (C-3’), 135.002 (C-1’’’), 134.741 (C-2’’), 132.649 (C-5’), 132.437 (C-1’’), 130.460 (C-3’’), 130.276 (C-4’’), 129.787 (C-4’’’), 129.421 (C-3’’’,5’’’), 128.925 (C-6’’,4’), 128.164 (C-2’’’,6’’’), 125.286 (C-5’’), 124.401 (C-6’), 124.283 (C-2), 122.342 (C-2’), 119.206 (C-5).
1-(3’-cinnamaamidophenyl)-3-(3’’-chlorophenyl)-prop-2-en-1-one (2i)
White (pale) colored solid, yield (75.86%). mp:165°C; FTIR (KBr, cm-1): 3321.81 (N-H stretch of anilide), 1664.57 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 761.88 (C-Cl stretch); 1H NMR (DMSO-d6, δ ppm): 10.595 (s, 1H, NH), 8.489 (s, 1H, H-2’), 8.206 (m, 2H, H-4’,2’’’), 8.121 (m, 2H, H-6’’’,3), 7.980 (d, 1H, H-6’), 7.898 (d, 1H, H-2), 7.797 (m, 3H, H-6, 5’,6’’), 7.724 (t, 1H, H-5’’), 7.674 (m, 5H, H-3’’’,4’’’,5’’’,4’’,2’’), 7.010 (d, 1H, H-5); 13C NMR δ ppm 189.405 (C-1), 164.194 (C-4), 142.771 (C-3), 140.998 (C-6), 140.158 (C-1’), 138.345 (C-3’,1’’), 137.302 (C-1’’’), 135.007 (C-3’’), 134.207 (C-5’), 131.110 (C-5’’), 130.583 (C-4’’), 130.280 (C-4’’’), 129.743 (C-4’), 129.430 (C-3’’’,5’’’), 128.368 (C-6’), 128.165 (C-2’’’,6’’’), 124.338 (C-2’’,6’’), 124.086 (C-2), 122.351 (C-2’), 119.220 (C-5).
1-(3’-cinnamaamidophenyl)-3-(4’’-chlorophenyl)-prop-2-en-1-one (2j)
Brown (pale) colored solid, yield (82.75%). mp:182°C; FTIR (KBr, cm-1): 3429.43 (N-H stretch of anilide), 1656.85 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 750.31 (C-Cl stretch); 1H NMR (DMSO-d6, δ ppm): 10.379 (s, 1H, NH), 8.212 (s, 1H, H-2’), 7.901 (d, 1H, H-4’), 7.787 (m, 4H, H-2’’’,6’’’,6’,3), 7.616 (d, 1H, H-2), 7.500 (m, 3H, H-6,2’’,6’’), 7.422 (m, 3H, H-5’,3’’,5’’), 7.326 (m, 3H, H-3’’’,4’’’,5’’’), 6.737 (d, 1H, H-5); 13C NMR δ ppm 189.432 (C-1), 164.227 (C-4), 143.027 (C-3), 140.921 (C-6), 140.205 (C-1’), 138.426 (C-3’), 135.535 (C-1’’’), 135.022 (C-5’), 134.009 (C-4’’), 130.916 (C-2’’,6’’), 130.260 (C-1’’), 129.723 (C-4’’’,4’), 129.411 (C-3’’’,5’’’,3’’,5’’), 128.163 (C-2’’’,6’’’), 124.235 (C-6’), 123.301 (C-2), 122.433 (C-2’), 119.224 (C-5).
1-(3’-cinnamaamidophenyl)-3-(3’’, 4’’-dichlorophenyl)-prop-2-en-1-one (2k)
White (pale) colored solid, yield (63.49%). mp:180°C; FTIR (KBr, cm-1): 3340.84 (N-H stretch of anilide), 3064.64 (aromatic C-H stretch), 1651.7 (C=O stretch of α, β unsaturated ketone), 750.08 (C-Cl stretch); 1H NMR (DMSO-d6, δ ppm): 10.472 (s, 1H, NH), 8.350 (s, 1H, H-2’), 8.069 (d, 1H, H-4’), 8.026 (m, 2H, H-2’’’,3), 7.904 (d, 1H, H-6’), 7.757 (m, 2H, H-6’’’,2), 7.661 (m, 4H, H-6,2’’,5’’,6’’), 7.592 (t, 1H, H-5’), 7.487 (m, 3H, H-3’’’,4’’’,5’’’), 6.874 (d, 1H, H-5); 13C NMR δ ppm 189.248 (C-1), 164.188 (C-4), 141.654 (C-3), 140.979 (C-6), 140.176 (C-1’), 138.269 (C-3’), 135.913 (C-1’’’), 135.006 (C-1’’), 133.159 (C-5’), 132.237 (C-3’’), 131.371 (C-4’’), 130.563 (C-4’’’,2’’), 130.252 (C-5’’), 129.699 (C-4’,6’’), 129.400 (C-3’’’,5’’’), 128.155 (C-2’’’,6’’’), 124.533 (C-6’), 124.383 (C-2), 122.357 (C-2’), 119.207 (C-5).
1-(3’-cinnamaamidophenyl)-3-(3’’-nitrophenyl)-prop-2-en-1-one (2l)
White (pale) colored solid, yield (80.53%). mp:205°C; FTIR (KBr, cm-1): 3317.56 (N-H stretch of anilide), 1662.64 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1514.12 (aromatic C=C stretch), 1346.31 (NO2 stretch); 1H NMR (DMSO-d6, δ ppm): 10.500 (s, 1H, NH), 8.801 (s, 1H, H-2’’), 8.383 (m, 2H, H-2’,4’’), 8.319 (d, 1H, H-4’), 8.146 (d, 1H, H-3), 8.085 (d, 1H, H-6’’), 8.041 (d, 1H, H-6’), 7.925 (d, 1H, H-2), 7.809 (t, 1H, H-5’’), 7.675 (m, 3H, H-2’’’,6’’’,6), 7.619 (t, 1H, H-5’), 7.503 (m, 3H, H-3’’’,4’’’,5’’’), 6.892 (d, 1H, H-5); 13C NMR δ ppm 189.382 (C-1), 166.270 (C-4), 148.921 (C-3’’), 142.051 (C-6,3), 140.243 (C-1’’), 138.212 (C-1’), 137.061 (C-3’), 135.503 (C-1’’’,6’’), 135.097 (C-5’), 132.286 (C-5’’), 130.864 (C-4’’’), 129.679 (C-4’), 128.935 (C-3’’’,5’’’), 128.223 (C-2’’’,6’’’), 125.684 (C-6’), 125.345 (C-2’’,4’’), 124.804 (C-2), 123.551 (C-2’), 120.673 (C-5).
1-(3’-cinnamaamidophenyl)-3-(4’’-nitrophenyl)-prop-2-en-1-one (2m)
Buff colored solid, yield (83.89%). mp:207°C; FTIR (KBr, cm-1): 3329.14 (N-H stretch of anilide), 1664.57 (C=O stretch of anilide), 1612.49 (C=O stretch of α, β unsaturated ketone), 1510.26 (aromatic C=C stretch), 1344.38 (NO2 stretch); 1H NMR (DMSO-d6, δ ppm): 10.320 (s, 1H, NH), 8.223 (s, 1H, H-2’), 8.159 (d, 2H, H-3’’,5’’), 8.025 (d, 2H, H-2’’,6’’), 7.928 (m, 2H, H-4’,3), 7.826 (d, 1H, H-6’), 7.707 (d, 1H, H-2), 7.501 (m, 3H, H-6, 2’’’,6’’’), 7.444 (t, 1H, H-5’’), 7.327 (m, 3H, H-3’’’,4’’’,5’’’), 6.716 (d, 1H, H-5); 13C NMR δ ppm 189.393 (C-1), 164.215 (C-4), 148.518 (C-4’’), 141.624 (C-3), 141.509 (C-6), 141.025 (C-1’’), 140.215 (C-1’), 138.137 (C-3’), 134.987 (C-1’’’,5’), 130.269 (C-4’’’), 130.209 (C-2’’,6’’), 129.813 (C-4’), 129.430 (C-3’’’,5’’’), 128.167 (C-2’’’,6’’’), 126.566 (C-6’), 124.508 (C-2), 124.355 (C-3’’,5’’), 122.322 (C-2’), 119.229 (C-5).
Biological Assay
Antiproliferative activity by Sulforhodamine B (SRB) assay
The in-vitro anticancer activity was measured for synthesized new compounds on breast cancer (MCF-7), chronic myelogenous leukemias (K562), glioblastoma (U373MG), and colon cancer (HT-29) using the Sulforhodamine-B stain (SRB) assay. The cell lines were grown in RPMI 1640 medium containing 10% fetal bovine serum and 2 mM L-glutamine. For the screening experiment, cells were inoculated into 96 well microtiter plates in 100 µL at plating densities as shown in the study details above, depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates were incubated at 37°C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental compounds. After 24 h, one 96 well plate containing 5×103cells/well was fixed in situ with trichloroacetic acid (TCA), to represent a measurement of the cell population at the time of drug addition (Tz). Experimental compounds were initially solubilized in DMSO at 100 mg/ml and diluted to 1 mg/ml using water and stored frozen prior to use. At the time of addition, an aliquot of frozen concentrate (1 mg/ml) was thawed and diluted to 10-6, 10-5, 10-4 and 10-3 μg/ml with complete medium containing test compounds. Aliquots of 10 µl of these different compound dilutions were added to the appropriate microtiter wells already containing 90 µl of medium, resulting in the required final concentrations i.e.10-7, 10-6, 10-5, 10-4 M. After the addition, plates were incubated at standard conditions for 48 h and assay was terminated by the addition of cold TCA. Cells were fixed in situ by the gentle addition of 50 µl of cold 30 % (w/v) TCA (final concentration, 10 % TCA) and incubated for 60 min at 4°C. The supernatant was discarded; plates were washed five times with tap water and air dried. SRB solution (50 µl) at 0.4 % (w/v) in 1 % acetic acid was added to each of the wells, and plates were incubated for 20 min at room temperature. After staining, unbound dye was recovered and the residual dye was removed by washing five times with 1 % acetic acid. The plates were air dried. Bound stain was subsequently eluted with 10 mM trizma base, and the absorbance was read on a plate reader at a wavelength of 540 nm with 690 nm reference wavelength. Percent growth was calculated on a plate-by-plate basis for the test wells relative to the control wells, and expressed as the ratio of average absorbance of the test wells to the average absorbance of the control wells × 100 [43, 44].
Using the six absorbance measurements [time zero (Tz), control growth (C), and test growth in the presence of drug at the four concentration levels (Ti)], the percentage growth was calculated at each of the drug concentration levels. Percentage growth inhibition was calculated as:
[(Ti-Tz)/(C-Tz)] x 100 for concentrations for which Ti>/=Tz (Ti-Tz) positive or zero
[(Ti-Tz)/Tz] x 100 for concentrations for which Ti<Tz. (Ti-Tz) negative
CDK2 and EGFR Kinase Inhibition Assay by ADP GloTM Assay
Kinase activities were performed using CDK2/CyclinA2 Kinase (Promega; Catalog #V2971), EGFR Kinase (Promega; Catalog #V3831), and ADP-Glo Kinase Assay kit (Promega; Catalog #V9101). Kinase enzyme kit contained Active enzyme, substrate, Reaction Buffer A (5X) and DTT solution (0.1M). Additionally, MnCl2 solution (2.5M) in EGFR kinase system. Reaction Buffer A (5X) was composed of 200mM Tris-HCl, pH 7. 5, 100mM MgCl2 and 0.5mg/ml BSA. ADP Glo kinase kit contained UltraPure ATP (10mM), ADP-Glo Reagent, Kinase Detection Buffer and Kinase Detection Substrate.
ADP-Glo Reagents were thawed at room temperature. Kinase Detection Reagent was prepared by mixing Kinase Detection Buffer with the Lyophilized Kinase Detection Substrate. Components of CDK2 and EGFR enzyme systems were thawed on ice. 2X buffer was prepared from Reaction Buffer A as given in protocol i.e. 1ml of 2X Buffer by combining 400μl Reaction Buffer A, 1μl DTT and 599μl of distilled H2O for CDK2 system. Similarly, 2X buffer was prepared from the Reaction Buffer A for EGFR system in the following manner:1 ml of 2X Buffer by combining 400μl Reaction Buffer A, 1μl DTT, 1.6μl MnCl2and 597.4μl of distilled H2O. Further, 1X buffer was prepared for both the systems by diluting corresponding 2X buffer with distilled water in 1:2 ratio. 1ml of 250μM ATP Assay Solution was prepared by adding 25μl ATP solution (10mM) to 500μl of 2X Buffer and 475μl of distilled H2O.
Reactions were performed in solid white 96-well polystyrene flat-bottomed plates in a final volume of 100µL. First step was to optimize the enzyme concentration. Different concentrations of enzymes were prepared in 1X Buffer. Reaction mixtures contained 10µL of diluted Active enzyme, 5µL of 1mg/ml stock solution of enzyme substrate ((Poly (Glu4, Tyr1) peptide substrate for EGFR system and Histone H1 substrate for CDK2 system) and 5µL of 2X Buffer. Further, 5μl of 250μM ATP assay solution was added in each well. After addition of ATP solution, the plates were incubated at 30°C for 15 minutes. The reactions were terminated by adding 25μl of ADP-Glo Reagent. Plates were shaken and then incubated for another 40 minutes at room temperature. After incubation, 50μl of the Kinase Detection Reagent was added, followed by incubation for another 30 minutes at room temperature. Luminescence was read using Microplate luminometer. The conversion curve (RLU vs log10[Enzyme concentration], ng) was plotted and corresponding signal-to-background ratio (SB) was calculated.
For screening of enzyme inhibitors, drugs were initially solubilized in dimethyl sulfoxide (DMSO) and diluted using distilled H2O. Three different concentrations were prepared viz. 1μM, 3μM and 10μM (concentration of DMSO in an assay does not exceed 2%). 5μL of different drug concentrations were added to the initial reaction mixture instead of 2X Buffer and reaction was carried out as done during enzyme optimization. Using the relative light units [RLUs] in the presence of drug at three different concentration levels, the percentage enzyme activity was calculated at each concentration of the compound tested. The IC50 values were calculated from a plot of the percentage enzyme activity vs. log concentration of the compounds [32, 45].
In-silico Computational Studies
Molecular docking
Molecular docking studies were done using Glide version 5.9 software [46, 47] in Schrodinger suite 2017-1. The crystal structure used for the studies were downloaded from protein data bank. These included PDB 2WXV for CDK2 [31] and PDB 1M17 for EGFR [48, 49]. These are complexed with inhibitor with ligand identifier WXV and AQ4; respectively. The protein structures were refined using the protein preparation wizard. All the water molecules that did not form any interactions or were not a part of the active site were deleted. Hydrogen atoms were added to the protein which included the protons necessary to define the correct ionization and tautomeric states of the amino acid residues. The protein structure was energy minimized using the impact refinement module. The steric clashes existing in the structures were removed using OPLS 2005 force field [50]. After protein prepartion, the receptor grids were generated using the receptor grid generation panel for CDK2 and EGFR, individually. The protein structures for CDK2 and EGFR were included as workspace entries and their ligands WXV and AQ4 were picked to be excluded from receptor grid generation using the ‘receptor’ tab. Default vdW scaling factor of 1.0 and charge scale factor of 0.25 were used. The centroid of co-crystallized ligand was used for the grid generation and the size for the ligands to be docked should be similar to the co-crystallized ligand. According to these specifications for the center and size the final grid was generated. No constraints were used [51]. The docking protocol was validated for both CDK2 and EGFR by removing the inhibitors from their complexes, re-docking, and calculating root mean square deviation (RMSD). The ligands were prepared using ligprep module [52] of Schordinger at pH 7.4 and then used for docking. The 2D ligand interaction diagrams for the docked complexes were visualized using the Maestro in Schrodinger suite 2016-4.
Molecular Dynamics Simulation
System Building
All molecular dynamics (MD) simulations were performed using the Desmond software [53, 54]. The initial coordinates for the MD simulations were taken from the docked complexes for compounds 2l and 2k with CDK2. The orthorhombic box with dimensions10 Å×10 Å ×10 Å approximately was used to apply the periodic boundary conditions. The TIP3P water molecules [55] were then added and the system was neutralized to balance the net charge of the system. Equilibration of the system was carried out using the default protocol provided in Desmond Software with OPLS2005 force field [56]. The equilibration consisted of a series of restrained minimizations that slowly relax the system without deviating substantially from the initial protein coordinates.
Simulation Details
Force field parameters for the protein–ligand systems were assigned using the OPLS-2005 force field. Heavy atom bond lengths with hydrogens and the internal geometry of water molecules were constrained using the SHAKE algorithm [57]. Periodic boundary conditions and a 9.0Å cut-off for non-bond interactions was used, with electrostatic interactions treated using the particle mesh ewald method. A default six-stage relaxation protocol was employed prior to the MD production run: (i) 2000 steps limited-memory Broyden–Fletcher–Goldfarb–Shanno (LBFGS) minimization (first 10 steps steepest descent algorithm) with the solute restrained and a loose convergence criterion of 50 kcal mol-1 Å-1; (ii) 2000 steps LBFGS minimization (first 10 steps steepest descent) with residues beyond 15 Å of ligands restrained and a convergence criterion of 5 kcal mol-1 Å-1. (iii) a short 12 ps simulation in the NVT ensemble using a temperature (T) of 10 K (thermostat relaxation constant = 0.1 ps) with non-hydrogen solute atoms restrained; (iv) a 12 ps simulation in the NPT ensemble using T = 10 K (thermostat relaxation constant = 0.1 ps) and pressure P = 1 atm (barostat relaxation constant= 50 ps) with non-hydrogen solute atoms restrained; (v) a 24 ps simulation in the NPT ensemble (T = 300 K; thermostat relaxation constant= 0.1 ps; P = 1 atm; barostat relaxation constant= 50.0 ps) with solute non-hydrogen atoms restrained; and (vi) a 24 ps simulation in the NPT ensemble (T = 300 K; thermostat relaxation constant = 0.1 ps; P = 1 atm; barostat relaxation constant= 2.0 ps) with residues beyond 15 Å of the ligands restrained. For all of the aforementioned atomic restraints, a 50 kcal mol-1 Å-1 2 restraint force constant was used, whereas target temperatures and pressures were controlled using Berendsen thermostat and barostat, respectively. For the dynamics, a multiple time step RESPA integration algorithm was used throughout, with time steps of 2, 2, and 6 fs for bonded, “near” non-bonded and “far” non-bonded interactions, respectively. Following the relaxation, a 20 ns MD run in the NPT ensemble was performed for each system using a Nose–Hoover thermostat and Martyna–Tobias–Klein barostat (T = 300 K, thermostat relaxation time = 1.0 ps; P = 1 atm; barostat relaxation time = 2.0 ps). Energy and trajectory atomic coordinate data were recorded at every 1.2 and 20.0 ps; respectively. An approximate number of 1000 frames were recorded [58, 59].
{kind=link}