General description of the data
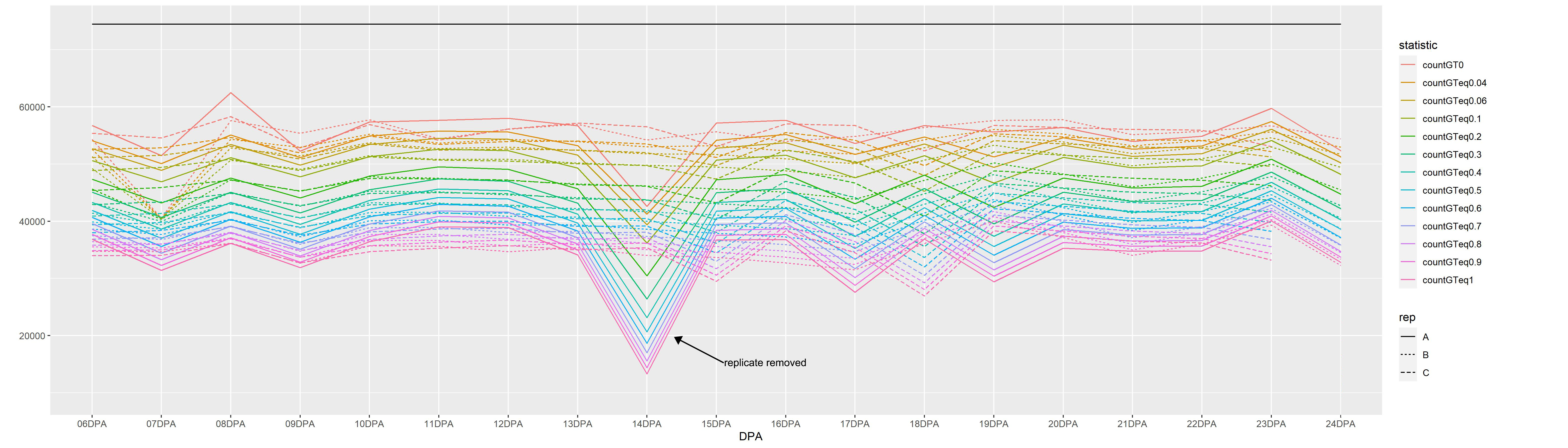
Gene expression during fiber development was surveyed from the early stages of PCW synthesis through the initiation and maintenance of SCW synthesis (i.e., 6–24 DPA; Fig. 1). Three replicates were collected for each stage, except 20 and 24 DPA where only two replicates were recovered. These samples yielded between 1.5 and 264.6 million (M) reads (mean = 41.2 M, median = 36.4 M) per sample. Clean reads were mapped to the 74,776 reference genes, resulting in an average of 34,020–55,008 genes expressed at any given time point for TPM > 0 and TPM ≥ 1, respectively (Supplementary Fig. 1; Supplementary Table 1).
Notably the number of expressed genes (~ 45 to 74% of transcriptome) is generally stable across replicates and DPA, with the exception of 14 DPA replicate A (Supplementary Fig. 1). Because 14 DPA replicate A had substantially fewer expressed genes than the other replicates (Supplementary Fig. 1), we removed this sample as a potentially early-aborted capsule.
Principal component analysis (PCA) of the expressed genes was used to explore patterns in the data (Fig. 2). In general, the first axis (PC1; 56% variance) clustered replicates and sequentially separated DPA along a temporal axis (from left to right). A small gap on the primary axis is observed between 9 and 10 DPA, which reflects the middle of elongation via PCW synthesis. Notably, the largest gap in the primary axis (PC1) is between 16 and 17 DPA, which is at the beginning of the transition stage (~ 16–20 DPA; [53]). Interestingly, the initial four timepoints (6–9 DPA) and last six timepoints (19–24 DPA) surveyed exhibited little differentiation along the primary axis, perhaps suggesting relative consistency in expression and/or tighter regulation of expression during the stages of early elongation and early CW thickening, respectively. The seven intervening timepoints (10–16 DPA), which are spread out along the primary axis, are correlated with the majority of elongation before the transition phase begins.
Gene expression trends across fiber development
Differential gene expression was evaluated for all 74,776 reference genes for adjacent stages, as summarized in Fig. 2. In general, the number of differentially expressed genes was equivalent between both subgenomes (i.e., AT and DT). Consistent with the aforementioned observation of a distinct difference between 16 and 17 DPA samples (Fig. 2), the number of differentially expressed genes (DEG) between those timepoints was more than an order of magnitude greater than most other comparisons (11,417 DEG, versus 16 − 5,562 in other comparison; median = 269 DEG, mean = 1,531 DEG; Supplementary Table 2), suggesting massive changes in gene expression correlated with entering the transition stage. Other, smaller spikes in DEG number were also apparent in the subsequent two comparisons (i.e., 18 versus 17 DPA and 19 versus 18 DPA), as well as between 22 and 23 DPA (Fig. 3). Few sharp increases were seen prior to the transition phase, save for small increases in DEG between 7–8 DPA and between 12–13 DPA. Interestingly, despite the disjunction between 9 and 10 DPA evident in the PCA plot, few genes exhibited significant differences in expression, suggesting that this apparent disjunction between these two DPA is the result of numerous subtle (i.e., not statistically significant) changes in gene expression.
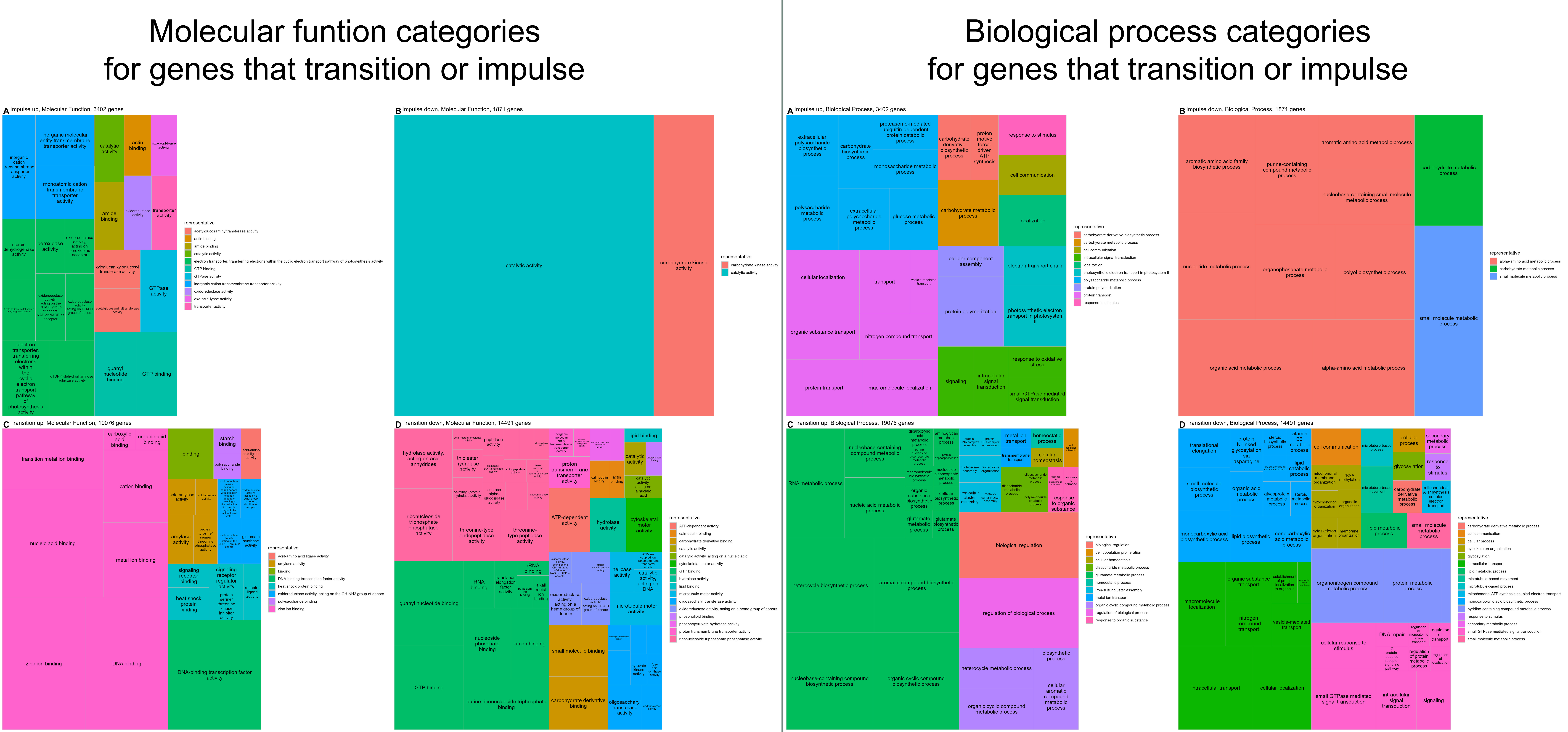
On average, the number of genes exhibiting down-regulation on adjacent days slightly out-numbered up-regulation (average of 805 versus 726, respectively) across the developmental timeline surveyed here. In nearly two-thirds of the adjacent DPA contrasts (60%; 11 contrasts), the number of down-regulated DEGs at the later days outnumbered the number of up-regulated DEGs; however, the opposite it true when evaluating patterns of differential expression in the context of a timeseries. When fit to a continuous model of gene-wise expression using ImpulseDE2, the number of genes that transition up (Tr-Up; 19,706 genes) or are transiently upregulated (Im-Up; 3,402 genes) during this developmental period (6 to 24 DPA) outnumbers those that transition down (Tr-Down; 14,491genes) or are transiently downregulated (Im-Down; 1,871 genes; Fig. 4). The broad classifications of genes in these categories are available in (Supplementary Fig. 2). As defined by ImpulseDE2 (see methods), genes in the transition categories either continuously increase (TrGene-Up) or decrease (TrGene-Down) their expression throughout the sampled time period. The 19,076 genes in the TrGene-Up category encode: glycoside hydrolases with a predicted role in deconstructing CW matrix polymers such as those found in the CFML [51]; transcriptional regulators of SCW synthesis; polysaccharide synthases, including cellulose synthases in all six major classes; accessory protein participants in cellulose synthesis; modulators of the microtubule and actin cytoskeleton; FASCICLIN-like arabinogalactan proteins; hormone response regulators (e.g auxin, brassinosteroid, ethylene, gibberellin, and jasmonic acid); producers and scavengers of reactive oxygen species; and many other proteins that can be logically associated with cotton fiber development (see other text and references in this article). The TrGene-Up category also includes homologs of many other regulatory and structural proteins that have been characterized in cotton or other species (primarily Arabidopsis), as well as many uncharacterized proteins.
The transient (or impulse) categories refer to genes whose expression profiles exhibit either increased (Im-Up) or decreased (Im-Down) expression during the middle of the time course and relatively lower or higher expression at the beginning and end, respectively. Interestingly, the beginning of the impulse periods (i.e., where Im-Up and Im-Down genes change expression) coincides with the small disjunction on the PCA between 9 and 10 DPA and the apex of the impulse period coincides with the major shift in gene expression between 16 and 17 DPA. The latter apex is particularly interesting, as it may reflect genes which regulate or participate in the massive changes in gene expression observed at the onset of the transition phase. Gene ontology (GO) analyses of these categories reveal many terms enriched within the Im-Up category for both Molecular Function (MF) and Biological Process (BP), and comparatively fewer terms for the Im-Down category (Supplementary Fig. 2). Among the Im-Up genes (i.e., *up; Fig. 4) are genes related to CW extensibility [4], which is required for rapid elongation. Interestingly, the proportion of transcription factors in the Im-Up category (10.1%) is significantly lower than found in the Tr-Up category (20.3%, p < 0.01 1-sample proportion test) and the proportion of Im-Down transcription factors (17.9%) is significantly greater than that found in the Tr-Down category (10.7%, p < 0.01 1-sample proportion test).
Interestingly, the time points sampled captured a small number of genes whose expression increased sharply between 23 and 24 DPA. DEG analysis revealed 201 genes upregulated at 24 DPA relative to 23 DPA (log2 fold change of 0.80–33.34), with 82% of the genes having log2 fold change > 2.0. Among these include genes that may be involved in the dominant process of cellulose deposition (see discussion) that begins circa 24 to 25 DPA in cotton fiber, including a GTPase protein (Gorai.011G031400, both homoeologs), two NAC transcription factors (Gorai.006G205300.A and Gorai.003G077700.D), and a MYB-like transcription factor (Gorai.001G138800.D).
Construction of a gene coexpression network
Expression relationships among genes were first analyzed using coexpression network analysis, which places genes into modules based on their correlated expression patterns and summarizes the expression of the genes within each module as the eigengene (i.e., the first principal component of the module). Approximately 7% (5,237) of the 74,446 genes were removed due to zero variance across the sampled times. The remaining 69,209 genes were placed in 18 modules, referred to as ME0 through ME17 (Fig. 5; Supplementary Fig. 3), where ME0 (9,748, 14.1%) comprises genes whose expression could not be assigned to a coexpression module [69]. Interestingly, the first two true modules (i.e., ME1 and ME2) each contain over 25% of the genes in the network. ME1 comprises 22,583 genes (32.6%) and exhibits an eigengene profile consistent with increased expression over time (Fig. 5; Supplementary Fig. 3). Intersection between ME1 and the Tr-Up category of differential expression (above) reveals 16,705 genes from ME1 are also contained within that category (Supplementary Table 3), representing 87.6% of the Tr-Up genes and 74.0% of ME1 genes. Complementing ME1, ME2 (18,919 genes; 27.3% of network) exhibits an eigengene profile consistent with decreasing expression over the time series. Similar to ME1, a majority of Tr-Down genes (12,991 genes, or 89.7%; Supplementary Table 3) are contained within ME2, comprising 68.7% of the total genes in ME2. Notably, the expression profiles of the eigengenes for these first two modules exhibit axial flips between 16 and 17 DPA (Supplementary Fig. 3), reflecting both the disjunction observed in the PCA and the major shift in gene expression exhibited in the time series differential expression analysis.
Both ME1 and ME2 also contain relatively high proportions of the Im-Up and Im-Down genes (Supplementary Table 3). ME1 contains 26.4% (899 out of 3,402) of the Im-Up genes and 21.4% of the Im-Down genes (400 out of 1,871), while ME2 contains 10.9% (370 genes) and 42.7% (798 genes), respectively. Although this represents 37.3–64.0% of genes contained within each impulse category, these genes represent only about 2–4% of the total genes in each module (Supplementary Table 3). While the expression trajectories of these transiently expressed/suppressed genes may not directly correspond to the module eigengene expression trajectory, their inclusion in these modules may indicate their participation in the general increase or decrease in expression of these modules.
The remaining modules (ME3 - ME17) contain far fewer genes (7,177 to 143, respectively), of which 12 modules are significant with respect to development (p < 0.05; anova ME ~ sample; Fig. 5). Notable among these are ME8 (766 genes) and ME9 (531 genes), both of which contain a relatively high proportion of the Im-Up genes (~ 13% each) relative to the remaining modules (except ME1; Supplementary Table 3). In both modules, more than half of the genes are assigned to the Im-Up category (ME8: 450 genes, 58.0% and ME9: 443 genes, 83.4%), which is reflected in their eigengenes, which start with low expression, peak in the middle of the timeseries, and then exhibit declining expression in the later time points; no Im-Down genes are detected in this category. This pattern is particularly apparent in ME9, which exhibits a sharp increase in expression between 9–12 DPA and a sharp decline between 16–19 DPA, and notably coincides with the fiber developmental periods encompassing rapid elongation and attenuation of elongation, respectively.
Three additional, consecutive modules (i.e., ME12-14) exhibit a high proportion of genes that are considered Im-Up or Im-Down (Supplementary Table 3), which is also somewhat consistent with their eigengene profiles (Fig. 5; Supplementary Fig. 3). Of those three, ME13 and ME14 have the greatest number of genes in the model that are Im-Up (i.e., 48.2% and 49.8% of module genes, versus 33.9% in ME12), and contain no genes that are considered Im-Down (as was observed for ME8 and ME9). Conversely, ME12 contains proportionately fewer Im-Up genes along with a small number of Im-Down genes (24; 6.1% of genes in module); however, the eigengene trajectory in ME13 is more similar to ME12 than it is to ME14. That is, both ME12 and ME13 exhibit an increase in eigengene expression starting around 13 DPA that subsequently plummets at ~ 23 DPA. ME14 exhibits a dissimilar profile (i.e., increasing steadily from 6 DPA followed by a sharp decline at 19 DPA) to both of these, suggesting that it may reflect a different aspect of fiber development.
With respect to the remainder of the Im-Down category genes, fewer modules (aside from ME1 and ME2) exhibit a relatively high number of these genes relative to the abundance in other modules (Supplementary Table 3). Interestingly, ME0 (i.e., unplaced genes) contains the third greatest number of Im-Down genes after ME1/ME2, perhaps indicating a role for some of these genes that is unclear from the current coexpression analysis. After ME0, ME4 and ME6 contain the most genes from the Im-Down category (ME4 = 157 and ME6 = 114), comprising 7.5% and 6.4% of the genes contained within each module, respectively. The eigengene for ME4 (Fig. 5, Supplementary Fig. 3) exhibits a transient-like pattern of expression, exhibiting a marked reduction between 13 and 18 DPA after which it sharply increases before tapering to 24 DPA. ME6, on the other hand, exhibits low expression until about 22 DPA, where it displays a sharp peak between 22 and 24 DPA, potentially indicating genes important for SCW synthesis, although the standard error for these DPA is high. Nevertheless, 243 genes from ME6 also exhibit significant DE between 22 and 23 DPA, most of which are classified as Tr-Up (226 genes). GO annotations for these genes are diverse, relating to metabolic processes (e.g., lipid, carbohydrate, and cellular), stimulus/stress response, etc.
Correlations between coexpression modules and measured phenotypes
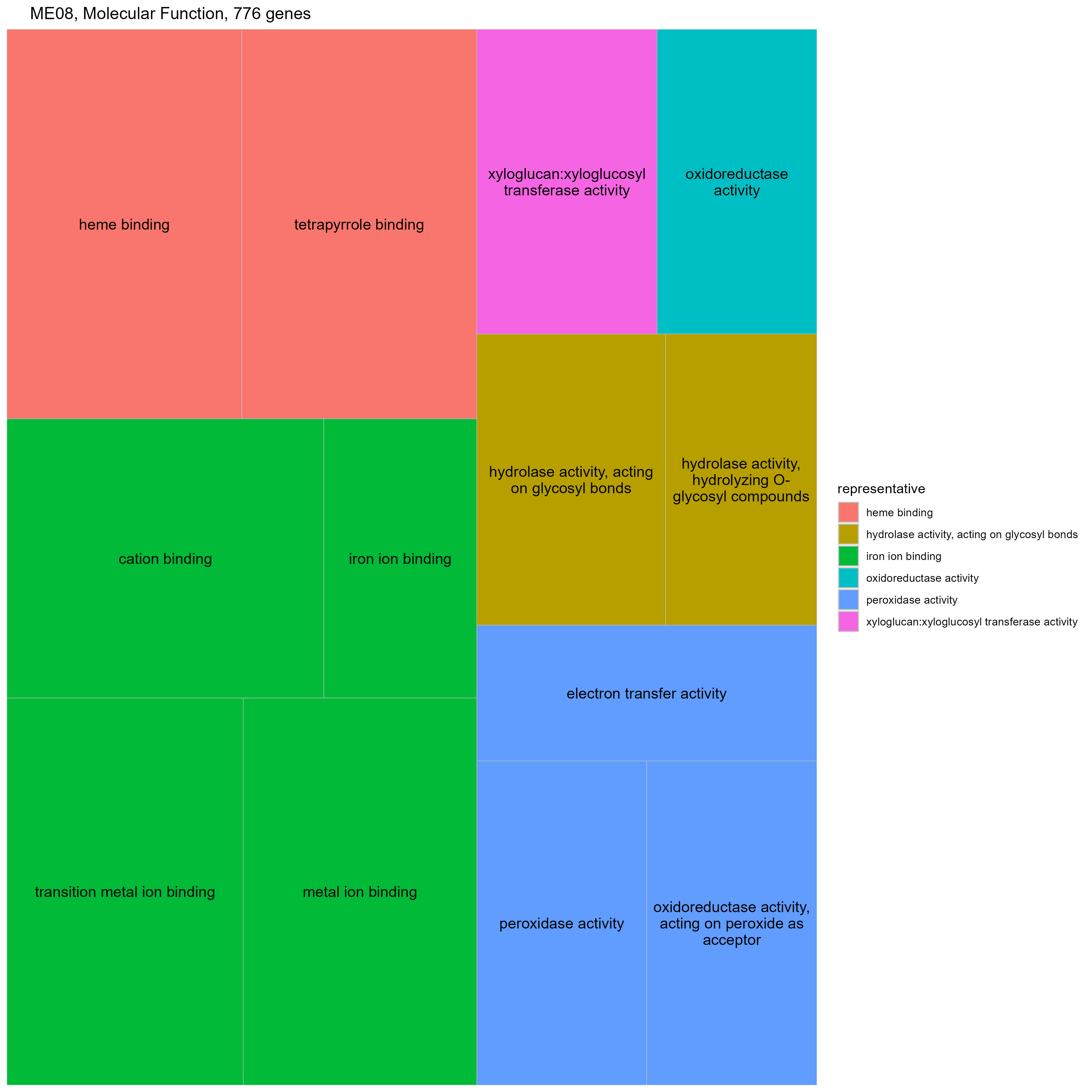
We correlated module eigengenes with phenotypic data gathered from the same accession (i.e., G. hirsutum cv TM-1) across the same developmental period (Fig. 6; Supplementary Table 4; [37]; Howell et al, in prep). As expected from the large number of genes present in the first two modules (22,583 and 18,919 genes, respectively) and the highly canalized nature of fiber development, most traits were significantly correlated (or inversely correlated) with those modules. Those molecules that contribute to CW development (e.g., encode genes involved in pectin, hemicellulose, and cellulose biosynthesis; [37]) were strongly positively correlated with ME1, which increases in expression over development and strongly negatively correlated with ME2, which decreases over time (Fig. 6). Likewise, fiber length (Howell et al, in prep) was strongly positively correlated with ME1 and negatively with ME2; however, these two traits also exhibit relatively strong, significant correlation with ME8 as well. As expected by the enrichment of Im-Up genes in this module, ME8 expression is impulse-like (Supplementary Fig. 3), whereby expression starts low, peaks at around 15 DPA, and then decreases again. GO analysis of the 776 genes in this module reveals glycosyl hydrolases, oxidoreductases, and peroxidases (Supplementary Fig. 4), which are all important for elongation.
Interestingly, turgor pressure exhibited strong correlations with different modules than the rest of the traits. Ruan and coworkers (Ruan et al 2001) used experimental data to estimate turgor values in 5, 10, 16, 20, and 30 DPA, which represented early, mid-, and late-elongation (5–16 DPA); transition or early SCW synthesis (20 DPA); and mid-SCW synthesis (30 DPA). For precise correlations with our daily transcriptome data, we interpolated the data to cover 6–24 DPA, which showed a gradual increase from 6–16 DPA (Supplementary Table 4). Although the interpolated data may be overly smoothed, there was a gradual increase to the peak at 16 DPA (0.67 MPa), followed by a decline through 20 DPA (0.28 mPa) and sustaining of similar values thereafter. Although turgor pressure is somewhat positively correlated with ME1 (r2 = 0.4) and negatively correlated with ME2 (r2=-0.48), stronger correlations were seen for ME8 (r2 = 0.9), followed by ME9 (r2 = 0.62) and ME13 (r2 = 0.54). Like ME8, ME9 and (to a lesser degree) ME13 exhibits impulse-like behavior, peaking between 13–16 DPA for ME9 and at 17 DPA for ME13. Although the present correlations leverage previously established turgor measurements for related (albeit nonidentical) materials, the strong correlations observed here suggest that evaluating turgor in the context of fiber development may provide additional insights into fiber development not typically captured by the standard fiber morphological measurements.
Construction of a crowd network
Because gene network inference algorithms are known to exhibit biases [70], we used Seidr [71] to generate a crowd network employing 13 algorithms (see methods), including the high performing GEne Network Inference with Ensemble of trees (GENIE3; [72, 73]) and Weighted Gene Coexpression Network Analysis (WGCNA). This network was aggregated using the inverse rank product [71, 74], resulting in 2.8 billion (B) edges (30%, or 0.85 B, “directed” edges) between all 74,446 nodes (genes) that exhibit variation among timepoints. Among these, 21,227 undirected and 15,996 directed edges connect nodes representing homoeologs. Since this dense network is composed of both “noisy” edges and those that represent core interactions, we calculated the network backbone to retain only those edges that represent the strongest connections for each node [71, 75]. Employing a 90% confidence interval reduced the number of edges over 500-fold to 5.1 million (M), which was further reduced to 2.2 M under a 95% confidence interval (see methods). Among these 2.2 M edges, the edge direction (i.e., which member of each pair of adjacent genes operates upstream of the other) is known for 721,101 edges (versus 1.5 M undirected edges). Despite the massively duplicated nature of this polyploid network, < 1% of surviving edges (10,761) connect homoeologs; however, just over half of those (5,422) of those are considered directed.
We compared these 2.2M backbone edges to the WGCNA-generated coexpression modules by first clustering the edges of the overall graph using two different algorithms, i.e., Louvain and InfoMap, which produced 188 and 1971 clusters, respectively. By overlapping these clusters with the WGCNA modules, we were able to place genes into 6,519 high confidence groups representing genes which are both placed within the same module and cluster using both algorithms. From these 6,519 groups, slightly less than half (3,094; or 47%) contain at least 1 edge (max: 118,932 edges and 2540 nodes), and possibly represent groups of genes that comprise small subdivisions of the broader gene network (Supplementary Table 5). As expected, the three largest clusters are derived from ME1 (1,500-2,540 genes each out of 22,583 genes total); however, the next largest clusters are not derived from ME1 or ME2 (module membership:~20k genes each) but are rather formed from genes placed in ME4 (1,438 out of 2,100 genes) and ME5 (1,298 out of 1,833 genes), the latter module which is notably not significant with respect to development. ME4, however, exhibits an eigengene profile consistent with Im-Down between 13 to 18 DPA, a pattern also consistent with the relative abundance of genes exhibiting transient down-regulation expression profiles. While the average and median number of genes per group is relatively low (16 and 3, respectively), 73 groups contain more than 100 genes (average = 424 genes; median = 214) connected by at least 113 edges (average = 11,408; median = 1,620).
Because gene regulatory networks provide insight into the regulatory hierarchies among genes, we isolated those 850 M edges representing the directed gene expression network from the broader crowd network for further analysis. From the top 10% of these edges (i.e., 8.5 M edges), few edges (7,330 or 0.09%) link homoeologs, most of which (5,422 or 74%) are retained in the network backbone described above. Louvain and Infomap clustering of these 8.5 M is similar to the above in that Infomap produces far more clusters (626) than Louvain (5); however, this clustering is notable in the small number of Louvain clusters (5), two of which together contain nearly 92% of genes (Louvain cluster 1 = 35,619 genes, or 48%; Louvain cluster 3 = 32,735 genes, or 44%). When the composition of these clusters is merged with each other and the module designations by WGCNA, it results in 2,206 cluster-groups (Louvain-Infomap-WGCNA), approximately one-third the number of cluster-groups in the backbone that includes both directed and undirected edges. These clusters (Supplementary Table 1; Supplementary Table 5) represent the most confident directed associations among genes in this dataset.
Phenotypic association between cellulose content and gene regulatory networks
Cellulose deposition in plant cells, including cotton fibers, is a tightly coordinated process driven by cellulose synthase complexes (CSC; [38]). Because mature cotton fibers are predominantly composed of cellulose, the orientation of cellulose microfibrils and the amount of cellulose deposited in the SCW are major determinants of key fiber properties (e.g., length and strength). As expected from the integral role of cellulose, crystalline cellulose accumulation (as measured in [37]) is significantly associated with nearly half (8) of the 17 coexpression modules (Fig. 6). Also as expected, cellulose accumulation is most strongly positively correlated with ME1 and most strongly negatively correlated with ME2, the two most gene-rich modules in the coexpression network; however, a strong positive correlation (0.65) was found with the 1,784 genes comprising ME6 and a strong negative correlation (-0.74) was found with the 283 genes comprising ME14. ME6 exhibits generally low expression until around 22 DPA, where it increases rapidly. This module (ME6) notably contains two CesA interacting genes (i.e., a KORRIGAN1-like, KOR1, and a COMPANION OF CELLULOSE SYNTHASE3-like, CC, gene; Gorai.003G089600.A and Gorai.005G256100.D, respectively), which are involved in cellulose synthesis [76].
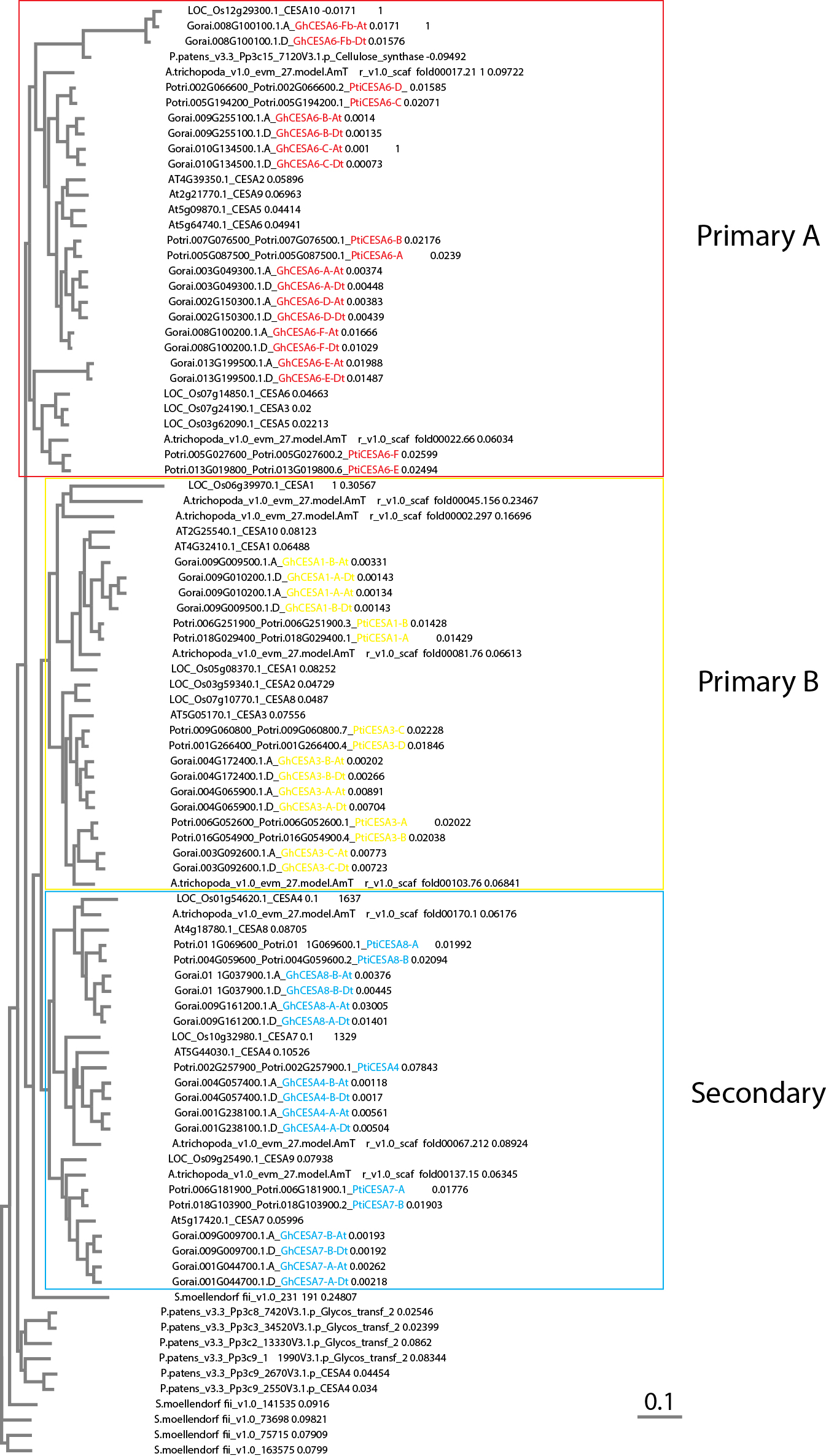
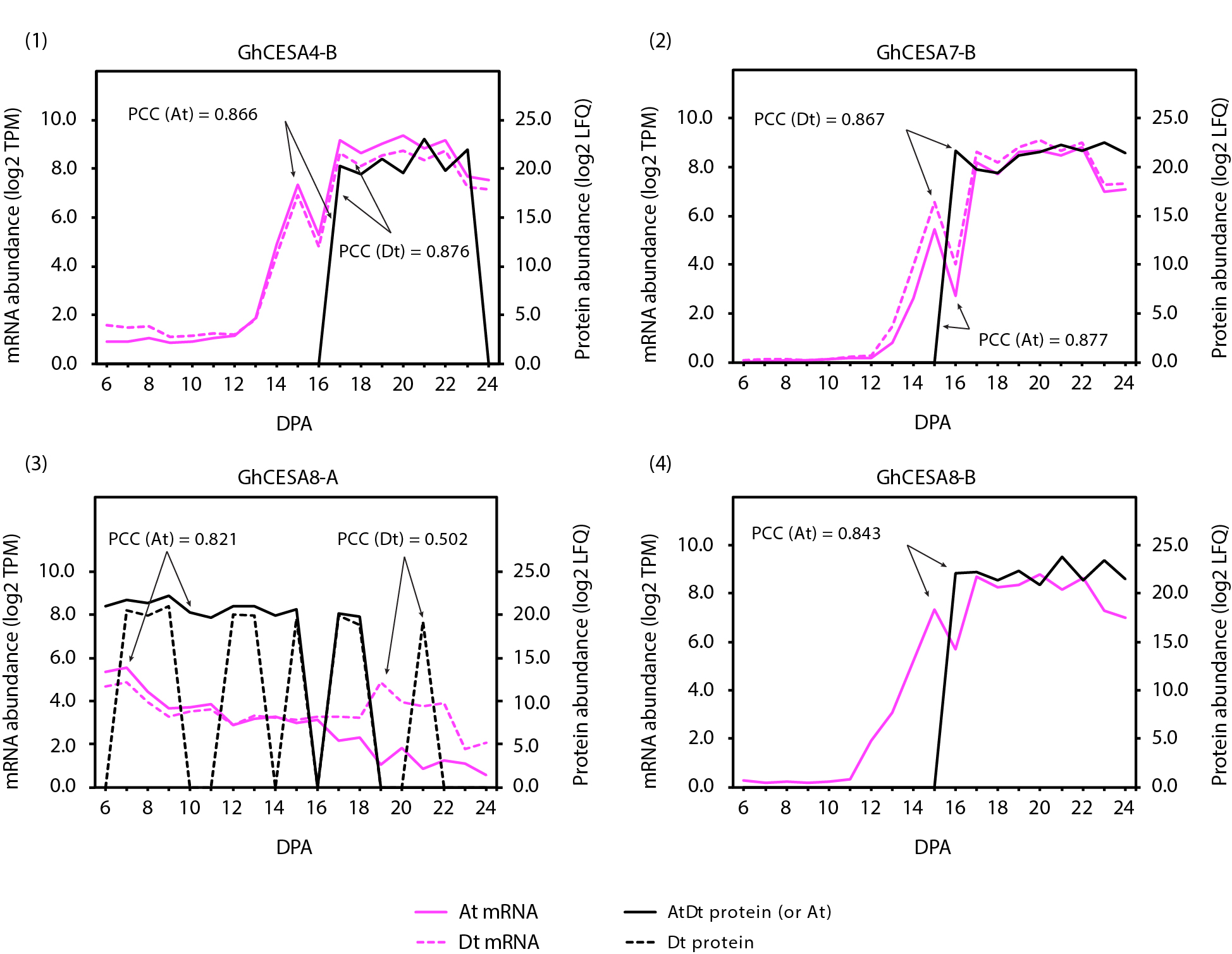
Phylogenetic analysis of the annotated cotton homoeologs with existing cellulose synthase A (CesA) homologs from Populus trichocarpa [77, 78] and other species revealed 24 G. hirsutum CesA genes related to PCW and 12 related to SCW (Supplementary Fig. 5; Supplementary Table 6). Due to strong conservation of CesA families in vascular plants, expression of CesA genes can be broadly partitioned into three major isoform classes each that are expressed during PCW (CesA1, 3, 6 or 6-like) or SCW synthesis (CesA4, 7, 8), assuming the 10-member CesA family of Arabidopsis as the canonical reference point [79]. Genome duplication in G. hirsutum has fostered expansion of the expression set for most of the major CesA classes, while also resulting in a non-canonical expression pattern during PCW synthesis for CesA8-A homologs. We observed that the three canonical PCW CesA classes typically maintain relatively even expression throughout, which may correlate with sampling ending early in SCW synthesis. In contrast, representatives of the three major SCW CesA gene classes, which co-function during SCW cellulose synthesis, are all expressed at a low-level beginning at 13 DPA followed by increasing expression during the transition stage and the onset of SCW synthesis. In an exception, there is a decrease in expression for both homoeologs of CesA8-A, as noted previously [53, 80], which may indicate that only the CesA8-B paralog fulfills the canonical role in SCW synthesis at DPA. In most cases (i.e., CesA7-B, CesA8-A, CesA8-B), the maternal and paternal homoeolog expression profiles were similar within gene; the sole outlier (Fig. 7), paralog CesA7-A (Gorai.001G04470), exhibited both comparatively reduced expression in the A homoeolog, as well as a delayed increase in expression (+ 5 DPA) that peaked at the same time as the rest of the SCW paralogs (~ 20 DPA).
We explored the gene-to-protein expression connection for these genes by comparing the abundance of CesA proteins in the membrane-associated (P200) fraction to the transcriptional data for the same DPA profiled here, which detected several secondary wall CESAs from fiber cell extracts (Fig. 7). Of the 36 CesA homoeologs, all but two (i.e., putative paralogs CesA6-E-At and Dt; see Supplementary Table 1) exhibited measurable gene expression (Fig. 7) and in all cases both homoeologs were distinguishable in the gene expression data. Due to the challenges of protein identification, however, only a subset of those genes were quantifiable via mass spectrometry (16, typically SCW-related; Fig. 7; Supplementary Fig. 6), most of which were ambiguous with respect to homoeolog of origin (all but CesA-8A and CesA-8B). All of the quantifiable proteins were derived from the membrane-associated (P200) fraction, which is expected due to the multiple transmembrane domains present in CesAs [81]. Notably, the demonstrated presence of CesA8 proteins during PCW synthesis points to the need for future research to understand their specific function at this time (see [82] for a review of less common potential roles for CesA8 orthologs in other species and tissues).
Overall, protein expression profiles for SCW cellulose synthase subunits were generally consistent with their corresponding gene expression profiles, albeit with approximately a 2–3 day difference in expression peaks (Fig. 7; Supplementary Fig. 6). Abundance profiles for GhCESA4-B, GhCESA7-A/B, and GhCESA8-B proteins were similar to their respective transcripts (Fig. 7), being first detected at ~ 16 DPA and exhibiting a 2–3 day lag relative to their transcripts. These preliminary results provide a foundation for further exploration of CesA transcript-protein associations during fiber development.
We further compared the expression among CesA isoforms by considering putative regulatory elements involved in CesA gene expression. Using only the directed edges from the Seidr crowd network, we found putative known transcription factors [83] for 7 genes (10 homoeologs), representing ~ 41% of expressed CesA genes (~ 30% of CesA homoeologs; Supplementary Table 7). For 6 of the 10 homoeologs, only one transcription factor was directly connected to that gene (3 each for PCW and SCW synthesis); however, for the remaining 4 homoeologs (3 SCW, 1 PCW), between 2–9 putative transcription factors of varying scores and ranks were directly connected to those genes. For the PCW CesA, putative transcription factors were found for the homoeologs GhCESA3-C-At and GhCESA3-C-Dt, although interestingly by transcription factors from different classes (Myb and ARF, respectively; Supplementary Table 7), both of which function in fiber development [57, 84]. The other two PCW genes (GhCESA3-B-Dt and GhCESA6-B-At) are putatively regulated by DOF (DNA-Binding with One Finger) transcription factors, the latter of which has multiple candidate transcription factors from diverse families (Supplementary Table 7). Slightly more putative regulators were found for the SCW genes, likely because the onset of PCW was not sampled here. A single putative TF regulator was associated with GhCESA4-A-At, GhCESA4-A-Dt, and GhCESA4-B-Dt, i.e., a TALE TF (Gorai.003G156000.D; Supplementary Table 7 [85, 86]), that rapidly increases in expression beginning around 10 DPA (Tr-Up). Putative regulators for the other subunits were found only for GhCESA7-B-Dt and GhCESA8-B (both homoeologs), each of which had more than one potential TF, sometimes from diverse families. GhCESA7-B-Dt, for example, was associated with 7 possible regulators, including one GATA, two Myb, three NAC, and one TALE TF, with the strongest association (highest ranked edge) connecting GhCESA7-B-Dt to the Myb Gorai.004G138300.D (Supplementary Table 7). Likewise, GhCesA8-B-Dt was associated with 5 possible regulators, including one Dof, two Myb, and two TALE TF, with the strongest association with the TALE Gorai.004G206600.A (Supplementary Table 7). For GhCesA8-B-At, however, there were only two candidate TF, both of which were from the C2H2 family and one of which (Gorai.008G178000.D) exhibited a stronger association.
To understand the position of the SCW cellulose synthase homologs in the context of the broader gene regulatory network (GRN), we explored a subset of the crowd network enriched for the strongest associations between those cellulose synthases and neighboring genes. This strict filtering criteria (see methods) resulted in three subnetworks, a main subnetwork containing representative homologs for each SCW cellulose synthase isoform (i.e., CesA4, CesA7, CesA8; Fig. 7, hereafter SCW subnetwork) and two smaller subnetworks that contained only the GhCesA4-A homoeologs or only GhCesA8-B-Dt, both of which were less strongly connected to the larger subnetwork, given our filtering criteria (Fig. 8). The large subnetwork contained 3 CESA7s, 2 CESA4s, and 1 CESA8, consistent with the cofunction of the encoded proteins in SCW cellulose synthesis. Both homoeologs of GhCESA4-B are adjacent and linked in the network, occupying a somewhat central location. Notably, some of the putative cellulose synthase transcription factors mentioned above were not present in this subnetwork, likely due to the limited strength of their connections. As expected, several genes that are closely linked to the SCW CesA genes have been previously noted for their importance to fiber development. For example, a FASCICLIN-like arabinogalactan (FLA) precursor is adjacent to GhCESA8-2-At, as is a KOR1-like protein, both of which have been associated with SCW synthesis, but with unproven specific roles so far [76]. Another FLA-like protein is proximal to GhCESA7-B-Dt, as are a pectin-lyase and a O-glycosyl hydrolase (GH17) gene, which likely encode enzymes participating in cleavage of CW polymers. Different genes appear adjacent to the A-genome homoeolog for GhCESA7-B, including a gene for xylan side-chain synthesis (GhXAT-2) and a microtubule-associated protein (MAP65-like), the latter of which also appears to be influenced by GhCESA7-A-Dt and both homoeologs of GhCESA4-B. In addition, both GhCESA4-B homoeologs are also linked to previously noted CW genes such as another FLA, a beta-6-tubulin (TUB6), and a reduced wall acetylation gene (GhRWA1-4). Each of these observations has relevance to CW thickening and other transition stage events, as discussed below.
Phenotypic association between turgor pressure and gene interactions
Although high turgor pressure is implicated in rapid elongation of cotton fibers [87–89], few genes have been identified that may contribute to changes in turgor during fiber development [88, 90]. Although we used previously established turgor measurements, we still find strong associations between turgor and several modules that exhibit transient expression patterns, which is perhaps unsurprising given the transient nature of high turgor pressure in driving fiber elongation. Estimated values for turgor pressure were most significantly associated with ME8 (Fig. 5) in which gene expression was highest at 15 DPA followed by a gradual decline through 24 DPA. A total of 776 genes are in ME8, including two with functional annotations related to turgor (i.e., a SWEET-like gene (Gorai.003G074400.D) and a PIP-like gene (Gorai.002G198900.D)), both with Im-Up expression patterns similar to the module. There were four genes with functional annotations related to turgor in ME9, which contained 531 genes. ME9 generally contains genes with high expression at 13–16 DPA followed by a sharp decline. These ME9 turgor-related genes are: a PIP-like gene (Gorai.006G181300.A), a SWEET-like gene (Gorai.003G074400.A), a SUT/SUC-like gene (Gorai.010G030700.A), and a bHLH transcription factor (Gorai.010G147100.A). As with the two ME8 genes, these genes are considered ImUp, exhibiting increased expression during the intermediate stages and often showing peak expression before 15 DPA when elongation begins to slow down (Fig. 9). Notably, the K + transporter GhKT1 (here, Gorai.012G142000.A and Gorai.012G142000.D) originally noted by Ruan et al (2001) was not found within either of these modules, but rather in ME2 where it exhibits expression that transitions down (considered Tr-Down by ImpulseDE2), congruent with observations in Ruan et al (2001). A different K + transporter was identified in ME8 (Gorai.009G292800) that was also classified as Tr-Down, and two additional K + transporters (Gorai.012G082500.A and Gorai.012G082500.D) were identified in ME9, although their expression trend was not described by ImpulseDE2. See discussion for further interpretation of these and other genes relevant to turgor from this module.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}