2.1. Culture growth and maintenance
F. circinatum FSP34 (CMW350) and all of its nursery isolates (Table 1) was obtained from the Fusarium collection of the Tree Protection Co-operative Programme, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, South Africa. The fungal isolates were routinely cultured on quarter-strength potato dextrose agar (¼ PDA) (Merck Group, Modderfontein GP, South Africa) for 7 days and kept in the dark at 25 ºC. For biofilm formation, a culture was flooded with 2 mL of phosphate-buffered saline (0.2 M PBS; 10 mM NaH2PO4, 10 mM Na2HPO4, 150 mM NaCl, pH 7.2) to obtain conidial cells. These cells were then counted in a Neubauer chamber and adjusted to the desired concentration before downstream analysis.
2.2. Extracellular vesicle isolation and purification
2.2.1 Isolation using ultracentrifugation
Isolation of EVs from planktonic and biofilm cells of F. circinatum FSP34 was performed using differential ultracentrifugation following a previous protocol with minor adjustments (Rodrigues et al., 2007). Briefly, conidia of F. circinatum were collected from 7-day-old cultures grown on ¼ PDA using 0.2 M PBS (pH 7.2). Conidia was inoculated into 200 mL of potato dextrose broth (PDB) to a final concentration of 1 X 106 spores/mL. To maintain the planktonic state of the fungus, spores were allowed to grow under shaking conditions using the orbital shaker (100 rpm, Shake-O-Mat, Labotec, South Africa) while biofilm formation assay was conducted as previously described (Ratsoma et al., 2024), the spores were allowed to grow at static conditions for 72 h. To begin with EV isolation, cells, and debris were removed from the media of planktonic and biofilms by sequential centrifugation steps at 4,000× g for 15 min and 15,000× g for 30 min at 4 °C. The resulting supernatant was then filtered using a 0.45 μm membrane filter (Merck Millipore) to further remove any remaining cells or debris. The Amicon ultrafiltration system (100-kDa MWCO, Millipore) was then used to concentrate the supernatants to a final volume of approximately 20 mL. One part of the concentrated supernatants was subjected to ultracentrifugation (Beckman Coulter, Brea, CA, USA) at 100 000× g for 1 h at 4 °C to collect the EVs. The pellets enriched in EVs were washed twice with PBS at 100 000× g for 1 h at 4° C and stored in PBS as aliquots at -80° C until further use.
2.2.2. Purification of EVs using size exclusion chromatography
For size-exclusion chromatography (SEC), FSP34 EVs were concentrated using the Amicon® Ultra-4 Centrifugal system (100-kDa pore size) and purified as described previously by Kunene et al., (2023 ). This involved application of a mixture containing EVs with a cell membrane-specific fluorescent lipophilic dye (FM4-64; Thermo Fisher Scientific, South Africa) which was incubated for 15 min in the dark at room temperature. After incubation, the sample was added to a 10 mL plastic syringe stuffed with nylon stocking at the tip and stacked with 10 mL sepharose CL-2B (Sigma-Aldrich, South Africa), equilibrated with PBS. The sample was then eluted with PBS to collect 30 sequential fractions of 0.5 mL in black microtiter plates (Greiner, South Africa). Fraction fluorescence (excitation at 560 nm, and emission at 734 nm) was measured immediately using a Spectra Max M2 plate reader (SpectraMax paradigm, Multimode detection platform). Fractions with fluorescence levels above 3.0 relative fluorescence units (RFU) were pooled together, as "EV signal" after measurement and stored in PBS as aliquots at -80 oC until further use.
2.2.3. Physical characterization of EVs
Purified EVs of F. circinatum FSP34 were spotted on carbon-coated grids for adsorption for 5 min. The exposed vesicles were then negatively stained with 1% (w/v) uranyl acetate for 3 min. Finally, the EVs were visualized using transmission electron microscopy (TEM; JOEL JEM 2100F, JOEL Ltd., Tokyo, Japan).EVs were visually analysed using scanning electron microscopy. For this purpose, sterile 65 mm petri dishes containing glass coverslips and 200 mL PDB were inoculated with 20 µL conidial cells (to a final concentration of 2 x 105 cells /mL) and statically incubated at RT for 7 days. Glass slides were then removed and flooded and rinsed with PBS prior to adding the pre-fixative solution containing 1 mL of 2.5% (v/v) glutaraldehyde (Merck, South Africa) / formaldehyde (Merck, South Africa). After another PBS rinse, biofilms were fixed with 1% osmium tetroxide for 1 h. Following a final PBS rinse, the fixed biofilms were dehydrated sequentially using a series of graded ethanol (i.e., 15 minutes rinses each in 1 mL of ethanol at concentrations of 30 %, 50%, 70%, 90%, and three rinses in absolute ethanol). The dehydrated samples were then treated with a 50:50 mixture of hexamethyldisilazane (HMDS) and absolute ethanol for 1 h, followed by a treatment with HMDS only, after which they were left to dry overnight. The glass slides were mounted on rectangle aluminium stubs and carbon coated for 15 min using Qourum Q150T ES sputter coater (Qourumtech, UK). The stubs were observed in a JEOL JSM 6490LV scanning electron microscope (GenTech Scientific Inc., Arcade, NY, USA).
Particle size distribution and concentration of EV signals were measured using NanoSight NS500 (Malvern Panalytical, UK). The samples were vortexed for 1 minute, and a 5 mL sample was diluted with 5 mL of PBS (dilution factor of 2). Immediately after, a test volume of 1.5 mL of each sample was injected into the NTA and analysed in triplicate (each run = 30 s video). PBS was treated as a blank. The videos were captured and analysed using NTA 3.3 Dev Build 3.3.104 (Malvern Panalytical, UK). The camera sensitivity and detection threshold were optimized per video, and the temperature was set at 22 °C.
2.3. EV protein content quantification
The protein content of EVs was quantified to act as EV concentrations using the QuntiPro-BCA assay Kit (Walker, 2002). Therefore, following manufacturers' guidelines 20 μL of EV sample was added to a 96-well microplate followed by 200 μL of BCA reagent (Merck, South Africa). The plate was then incubated in the dark for 2 h at 37 °C. Post-incubation absorbance of the plate was measured at 560 nm and protein concentration was determined from a bovine serum albumin (BSA) standard curve.
2.4. Uptake analysis of biofilm derived-EVs by planktonic cells
2.4.1. EV membrane staining
The labelling of the biofilm derived-EVs (bEVs) was performed according to Regente et al., (2017). Briefly, purified vesicles were suspended in 40 μL of PBS and mixed gently with FM4–64 (Molecular Probes, Thermo Fisher Scientific, Argentina) to a final concentration 1 μg/mL and kept in the dark for 60 min on ice. After which, labelled samples were diluted with 3 mL PBS and ultracentrifuged at 100 000 x g to remove excessive dye, this was performed twice. The obtained pellet finally resuspended in 20 μL PBS.
2.4.2. Uptake of biofilm derived-EVs by FSP34 planktonic cells
A conidial suspension of F. circinatum adjusted to about 10 000 cells was incubated on a glass slide with 2 μL of bEVs labelled with 5 μg/mL of FM4–64. The cells were then visualized under confocal microscopy after 5 min of incubation at room temperature. Control treatments were performed by incubating conidia with PBS instead of FM4–64 labelled EVs. Following this, a microscopic examination of the cells was performed using a 63x oil immersion lens (ZEISS, CSLM), with FM4–64 excited at 488 nm and detected at 650-750 nm.
2.4.3. EV add-back assay of biofilm-derived EVs on FSP34 planktonic cells
Conidial suspension containing about 1500 cells was incubated with a mixture of 5 μL of purified bEVs (5 μg/mL) and 4% sucrose to a final volume of 20 μL. After 16 h of incubation at 25 °C, 5 μL of the mixture was evaluated for the presence and morphology of hyphae using a light microscope (LM, ZEISS, South Africa). Controls were performed by replacing EVs with the same volume of PBS. The experiment was performed in triplicate and repeated on different days. To perform the germ tube presence and cell viability analysis,the conidial suspension was co-incubatedwith bEVs as before, but this time around the cells were incubated for up to 24 h shaking at 25 °C in the dark. During the incubation period, cells were analysed every 24 h and at each time point, 20 μL of the cells were stained with tryphan blue (Merk, South Africa) and assessed for germ tube formation and cell viability. This assay was performed in triplicates using the countess 3FL cell counter (Invitrogen, ThermoFisher Scientific).
2.4.4. The effects of bEVs on pre-formed FSP34 biofilms
For this experiment, 72 h pre-formed biofilms of FSP34 were exposed to 5 μg/mL of either bEVs or heat-treated bEVs of FSP34 and incubated for 24 h. Post incubation, we quantified biomass, extracellular matrix (ECM), and metabolic activities by performing crystal violet, safranin assay, and XTT reduction assay, respectively with minor modifications, according to Ratsoma et al., (2024). Conidial suspension without bEVs was considered a positive control while the negative controls included conidial suspension with heat-treated (90 °C for 15 min) bEVs as well as ¼ PDB.
2.4.5. The effects of EVs purified from FSP34 heat-treated biofilm cultures on thebiofilm of nursery field isolates
*Fusarium circinatum isolates treated with vesicles released by heat treated biofilms (h-bEVs).
EVs were also purified from 72 h- old FSP34 biofilms that were subjected to heat at 45 ºC for an hour using SEC, as previously described. The FSP34 strain was then considered as an EV donor isolate. A total of 10 nursery field isolates of F. circinatum originating from diseased Pinus spp. trees found in South Africa were obtained from the Fusarium culture collection (CMWF) maintained at the Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, South Africa (Coutinho et al., 2007, Steenkamp et al., 2014) (Table 1). Among these, strains CMWF2597, CMWF2625, CMWF535, and CMWF568 were selected based on their antifungal response profiles to be recipients of EVs in order to understand the effects of EVs in promoting biofilm integrity and antifungal resistance. The source of EVs was prepared by forming FSP34 biofilms for 72 h under normal conditions (stationary at RT) and stressful conditions (72 h pre-formed biofilm) followed by 1 h exposure to heat at 45 ºC (Ratsoma et al., 2024). EVs from these sources were then purified using SEC as before and 5 µg/ml of these bEVs were applied to 72 h recipient biofilms formed by nursery isolates on microtiter plates to assess their impact on biofilm fortification.
2.5. The effect of bEVs on mono-and polymicrobial biofilm of nursery field isolates
Monomicrobial biofilms were established by inoculating each conidial suspension from individual isolates, while for polymicrobial biofilms made up of two isolates were co-incubated to a final concentration of 2 × 105 cells/mL in 96‐well flat‐bottom polystyrene plates containing 200 μL of PDB. At 72 h, pre-formed mono- and polymicrobial biofilms of nursery field isolates were exposed to 5 μg/mL of bEVs from FSP34 and incubated for 24 h. Post incubation, we quantified biomass, ECM, and metabolic activities by performing crystal violet, safranin assay, and XTT reduction assay, respectively with minor modifications, according to Ratsoma et al., (2024). Pre-formed mono- and polymicrobial biofilms without bEVs were considered a positive control while the negative control contained ¼ PDB only.
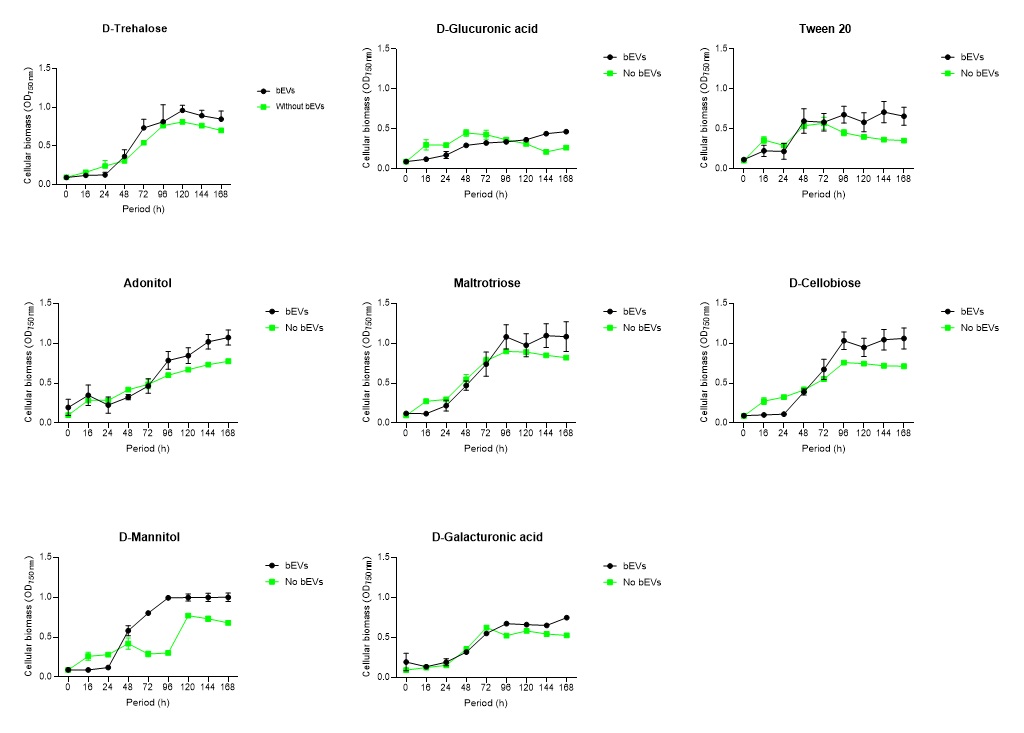
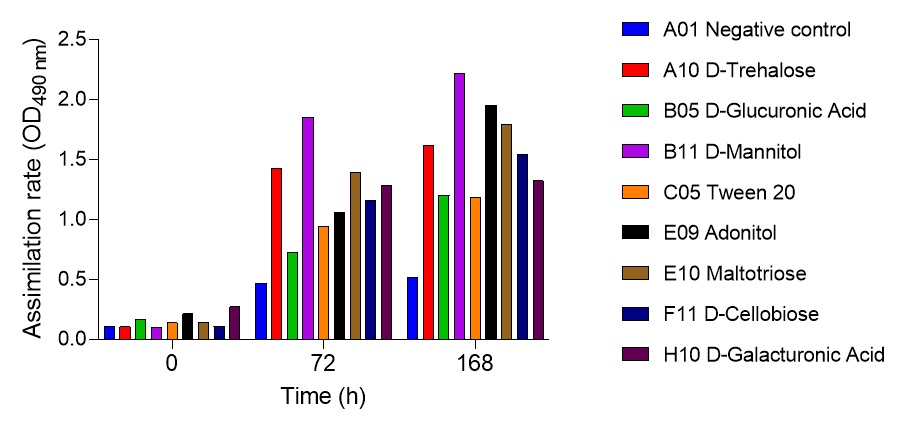
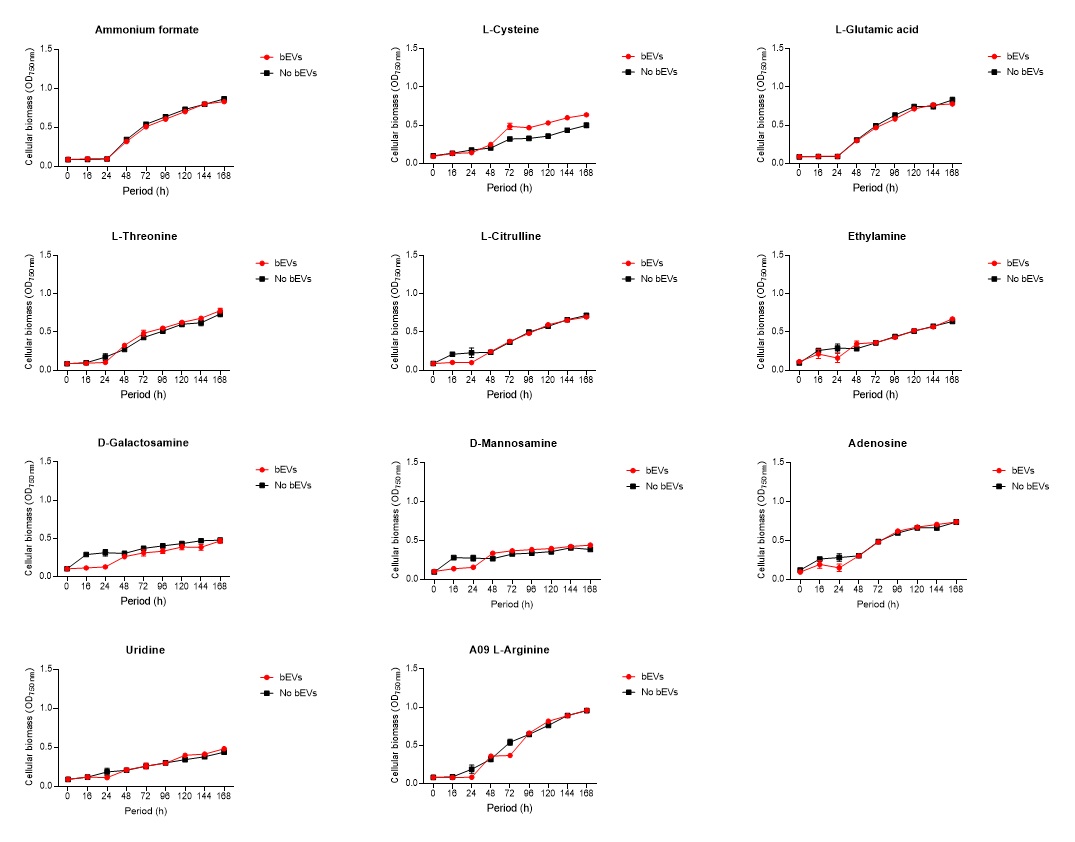
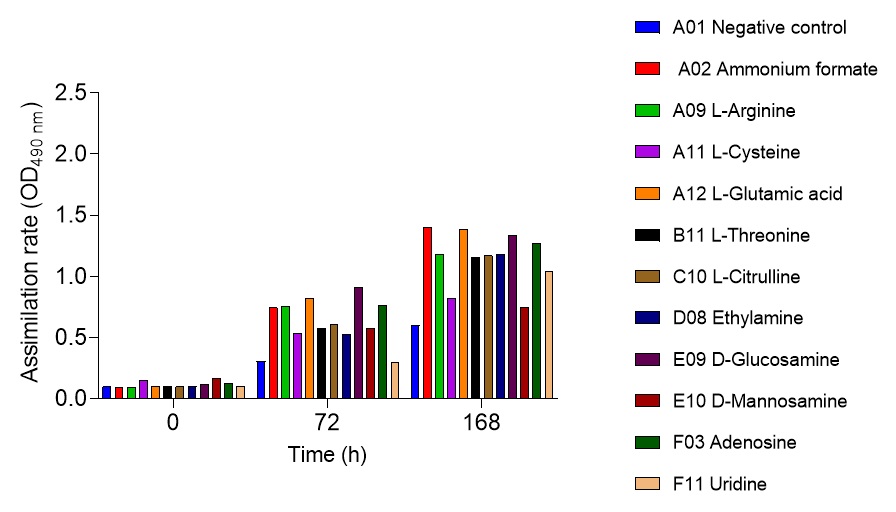
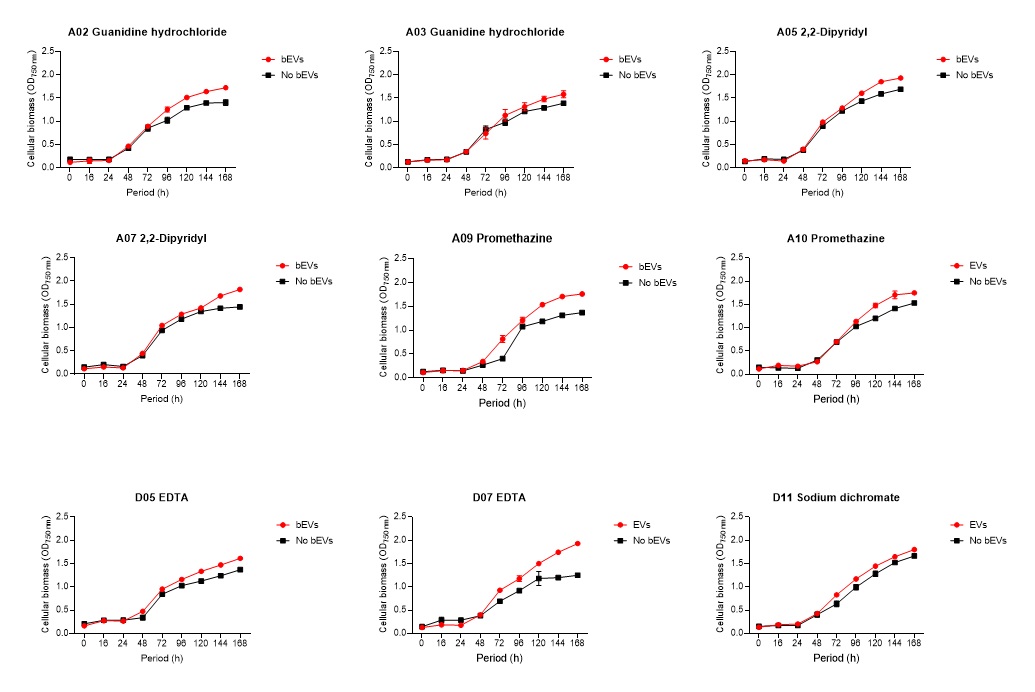
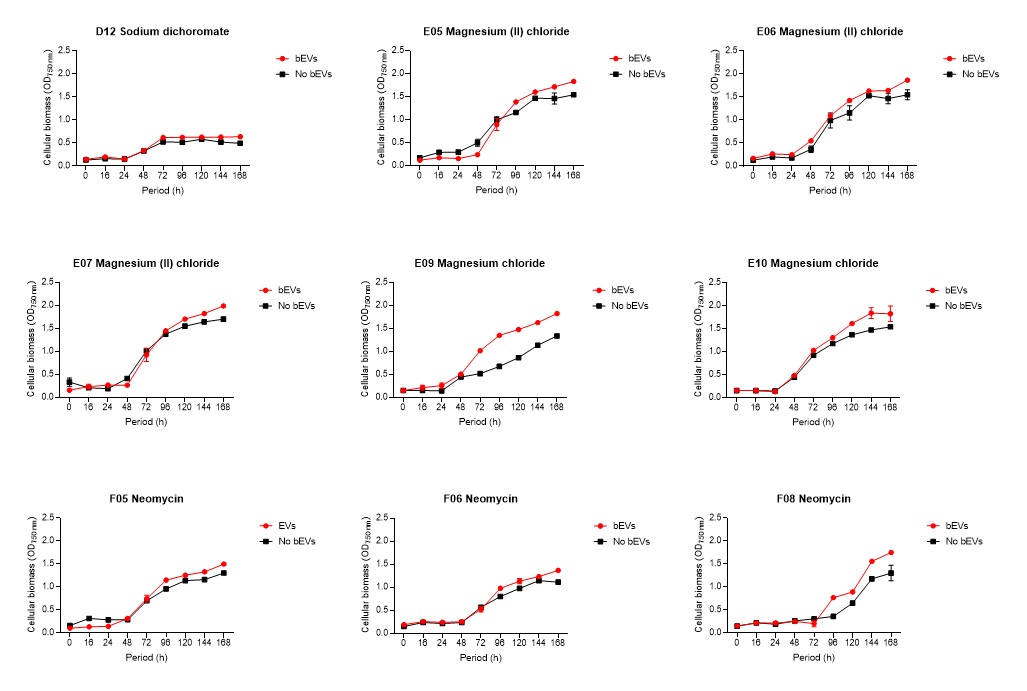
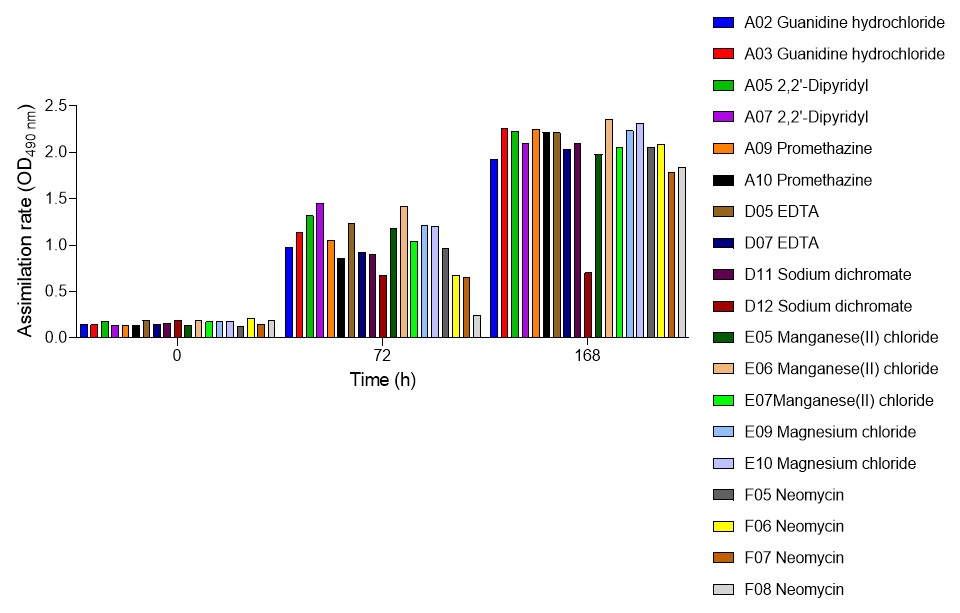
2.6. Global nutrient profiling of FSP34 spores treated with biofilm-derived EVs
F. circinatum utilization and assimilation profiles were generated for the FSP34 strain using the Filamentous fungi (FF) MicroPlates (Biolog®), namely, PM1, PM3B, and PM21D (Anatech, South Africa), respectively. For this analysis, we investigated the growth and metabolism of planktonic cells following co-incubation with bEVs. Each panel of the FF microplates contains 96 wells of which 95 wells represent specific carbon and nitrogen sources, while the one well contained water representing a negative control (Table S1). For chemical sensitivity analysis, each panel of the 96 well FF plate represented 24 different chemical agents (Table S1). For inoculum preparation, sterile swabs were soaked in PBS and then gently rolled over the mycelia of 7-day-old cultures. The spores on the swabs were then resuspended in 12 ml of FF inoculation fluid (FF-IF) (Anatech, South Africa) and mixed gently. The transmittance of this suspension was adjusted to 62% (0.1) using the spectrophotometer (SpectraMax paradigm, Multimode detection platform) at 590 nm. The FF inoculation fluid (IF) was prepared as shown in Table S1. A hundred microliters of FF-IF mixture were added to each well and incubated at 25 oC in the dark for 7 days. FF-IF mixture with bEV-treated spores was considered a positive treatment while the mixture with spores without bEVs was considered non-treatment (negative control), where PBS was substituted for bEVs. This experiment was performed in triplicate and readings were taken every 24 h for 7 days. Growth was measured using the spectrophotometer at an optical density (OD) of 750 nm to assess the cellular biomass (Tanzer et al., 2003).
2.7. Reproducibility and statistical analyses
Data was presented as mean ± standard error of the mean (SE). Statistical analyses were performed using GraphPad statistical software (GraphPad 8 Software, San Diego, CA, USA). The principal component analysis (PCA) plots were performed using R studios 4. 3 .2. For statistical analysis of viable counts, cultures containing 2 x 105 and 1 X 106, etc. CFU/ mL, were respectively used for EV isolation, biofilm formation and uptake analysis. All experiments were performed in triplicates.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}