2.1. Human tissue collection
A total of 74 cases of formalin-fixed paraffin-embedded samples, including 34 OC tissue specimens, 20 normal ovarian tissue specimens and 20 metastatic tissues, were collected from OC patients who underwent curative-intent surgery without prior radiotherapy and chemotherapy between 2014 and 2018 at the Department of Pathology, Xiangya Hospital of Central South University (CSU). All specimen diagnoses were confirmed by a qualified pathologist after surgery. The study was approved by the Institutional Ethics Committee of Xiangya Hospital of CSU and was conducted following the ethical guidelines of the Declaration of Helsinki. Each participant signed an informed consent form.
2.2. Cell culture
The epithelial OC cell lines HO-8910 and the monocytic cell line THP-1 were obtained from the Chinese Academy of Sciences Cell Bank and had been purchased from China Center for Type Culture Collection (CCTCC) and Cell Resource Center, IBMS, CAMS/PUMC (CRC/PUMC). HO-8910 cells stably overexpressing miR-205 and negative control cells were established by the Xiaoying Wu research group of the pathology laboratory of CSU, and the transfection efficiency was verified in a previous study [23]. All cell lines were cultured in DMEM or RPMI 1640 medium (Thermo Scientific, Waltham, MA, USA) supplemented with 10% foetal bovine serum (FBS) (Gibco, Gaithersburg, MD, USA). THP-1 cells (1×106) were incubated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA) (Solarbio, Beijing, China) for 24–48 h in vitro to induce their differentiation into macrophages. For the exosome treatment experiments, 1 µg/ml exosomes was added to the culture medium of recipient cells (2 × 105).
2.3. Immunohistochemistry (IHC) and in situ hybridization (ISH)
A total of 74 paraffin-embedded OC tissues were assessed by IHC and ISH staining. The macrophage marker CD68 and the M2-type macrophage marker CD163 expression were measured with anti-CD68 (1:200, Abcam, Cambridge, UK) and anti-CD163 (1:200, Abcam) antibodies. ISH was performed on OC tissue samples using an LNA microRNA ISH miR-205 optimization kit (Exiqon; Woburn, MA, USA). The stained slides were scanned and photographed using digital pathological scanning equipment. The final score obtained from the Image-Pro plus (IPP, version 5.0, Media Cybernetics, Silver Spring, MD) analysis was used to identify the relative level of CD68, CD163 and miR-205 expression. The positivity rate was calculated as the integral optical density (IOD)/area sum. The experimental steps of IHC and ISH were the same as in our previous study [24].
2.4. Exosome isolation and characterization
The cell lines were cultured in normal medium until 60–70% confluence, thereafter, the medium was replaced with fresh medium supplemented with 10% exosome-depleted FBS. Then, the cell culture medium was harvested after 48 h and centrifuged (Beckman Coulter, Brea, CA, USA) at 2,000×g for 30 min at 4°C to remove residual cells, debris and remaining macropolymers. The supernatant was passed through a 0.22-µm filter (Millipore, Danvers, MA, USA). After centrifugation at 3,000×g for 30 min at 4°C in a dialysis tube (Millipore), the supernatant was incubated with ExoQuick-TC™ exosome precipitation solution (System Biosciences, Palo Alto, CA, USA) for 6 h to overnight at 4°C. Subsequently, the mixture was centrifuged at 1,500×g for 30 min at 4°C to harvest the yellow exosome pellets. The exosome pellets were resuspended in 100 µl phosphate-buffered saline (PBS) for further assays. Western blotting analysis was used to characterize the extracellular vesicle-associated protein markers Hsp70 (Abcam) and CD63 (Abcam) and the exosome-specific marker TSG101 (Abcam). For transmission electron microscopy (TEM), the exosomes were fixed with 1% glutaraldehyde, dropped in 300-mesh carbon/formvar-coated grids, stained with 2% uranyl acetate, dried and imaged by transmission electron microscopy (TEM) (FEI, Hillsboro, OR, USA), as previously reported [26]. The amount of exosomes was measured using the bicinchoninic acid (BCA) protein assay kit (Novagen, Merck Group, Madison, USA).
2.5. Exosome labelling and tracking
To monitor exosome trafficking, purified exosomes isolated from the culture medium were collected and labelled with PKH67 red fluorescent membrane linker dye (Sigma-Aldrich, Merck KGaA) according to the manufacturer's instructions. Next, the PKH26-labelled exosomes were washed and centrifuged at 100,000×g for 20 min at 4°C to collect the exosomes, which were resuspended and added to unstained macrophages for exosome uptake studies. After treatment with PKH26-labelled exosomes for 4 h at 37°C, the cells were detected by fluorescence microscopy.
2.6. RNA interference, vector transfection and qRT-PCR
The miR-205 mimic (10 nM), inhibitor (20 nM), negative control (10 nM) and PTEN expression plasmid (2 µg, GeneCopoeia) were transfected into cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol. The cells were collected for further assays at 24 or 48 h after transfection. The transfection efficiencies were verified by fluorescence microscopy and RT-PCR analysis. Total RNA from cells and exosomes was extracted using TRIzol reagent (Invitrogen), and complementary DNA was synthesized with a reverse transcription system (GeneCopoeia, Guangzhou, China) according to the manufacturer's instructions. Reverse transcription and qRT-PCR were performed as previously described [26]. The relative expression levels were evaluated using the ΔΔCt method. The 2−ΔΔCt method was applied to analyse the results. The primers for miR-205 mimic, miR-205 inhibitor, miR-205 nc, CD163, CD68, IL10, CCL8, IL-1β, TNF-α and PTEN (GeneCopoeia) are available in the Supplementary Table 1.
2.7. Plasmids, transient transfections and luciferase assays
An miR-205 promoter (420 bp) was amplified from the mouse genomic DNA template and inserted into a pGL3 vector (Promega Corporation, Madison, WI, USA). Reporter vectors carrying the miR-205 target site were constructed by synthesizing a 3’-untranslated region (UTR) fragment containing the predicted target sites (miRWalk) for PTEN cDNA, and subsequently inserting the PTEN cDNA fragment into the multiple cloning site of a pMIR-REPORT™ luciferase miRNA expression reporter vector (Ambion Life Technologies, Carlsbad, CA, USA). For the luciferase reporter assay, HO-8910 cells were seeded in 24-well plates and transfected with the indicated plasmids. Cells were harvested 36 h after transfection. Luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Promega Corporation).
2.8. Western blot analysis
Proteins extracted from cells or exosomes were lysed in RIPA lysis buffer supplemented with protease inhibitor and quantified using a BCA protein assay kit (Novagen, Merck Group, Madison, USA). Approximately 30 µg of protein lysates (from each sample) were separated on 10% SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 5% blocking buffer overnight at 4°C with primary antibodies, followed by incubation with a secondary antibody at room temperature. The primary antibodies included anti-CD63, anti-TSG101, anti-Hsp70, anti-E-cadherin, anti-vimentin, anti-PTEN, anti-mTOR, anti-p-mTOR, anti-AKT, anti-p-AKT, anti-4EBP1, anti-p-4EBP1 (Cell Signaling Technology, USA) and anti-α-tubulin (Proteintech, Chicago, USA). The chemiluminescence signal was detected using an enhanced chemiluminescence mixture (Sigma-Aldrich), and images were captured using a gel imaging system (Bio-Rad Laboratories, CA, USA).
2.9. Co-culture, cell migration and invasion assays
Before starting the assays, THP-1 cells (1×104) were seeded on the bottom chamber of 8-mm Transwell inserts (Corning, Sigma-Aldrich) with PMA (100 ng/ml) for 48 h. Then, the THP-1 cells were differentiated into M0 macrophages, and the supernatant was replaced with normal medium containing 10% FBS for 24 h to eliminate the effect of PMA. Tumour cells (5×105) were seeded on the upper side of the co-culture system before the co-culture assay. After treatment with miR-205-Exos or miR-205 mimic/NC/inhibitor (RiboBio, Guangzhou, China), M0 macrophages were co-cultured with HO-8910 cells. Cell migration and invasion assays were performed as previously described [23].
2.10. Animal experiments
All animal experiments were performed in accordance with the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals and according to the protocols approved by the Animal Care and Use Committee at CSU. Female BALB/c nude mice (4–5 weeks old, 18 ~ 20 g) were housed in a specific pathogen-free environment in the Animal Laboratory Unit of CSU and were randomly divided into two groups (n = 6/group). HO-8910 cells (1.5×106 cells) mixed with conditioned macrophages pretreated with NC-Exos or miR-205-Exos were injected intraperitoneally (i.p.) into different groups. The procedure of living animal bioluminescence imaging was described previously [24].
2.11. Statistical analysis
All experiments were performed in triplicate. Data are presented as the mean ± standard error of the mean (SEM). Differences between treated and control groups were analysed using Student’s t test or one-way ANOVA. Survival curves were estimated using the Kaplan-Meier method, and the log-rank test was used to calculate differences between the curves [25]. A P-value less than 0.05 was considered statistically significant. Statistical analyses were performed by GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
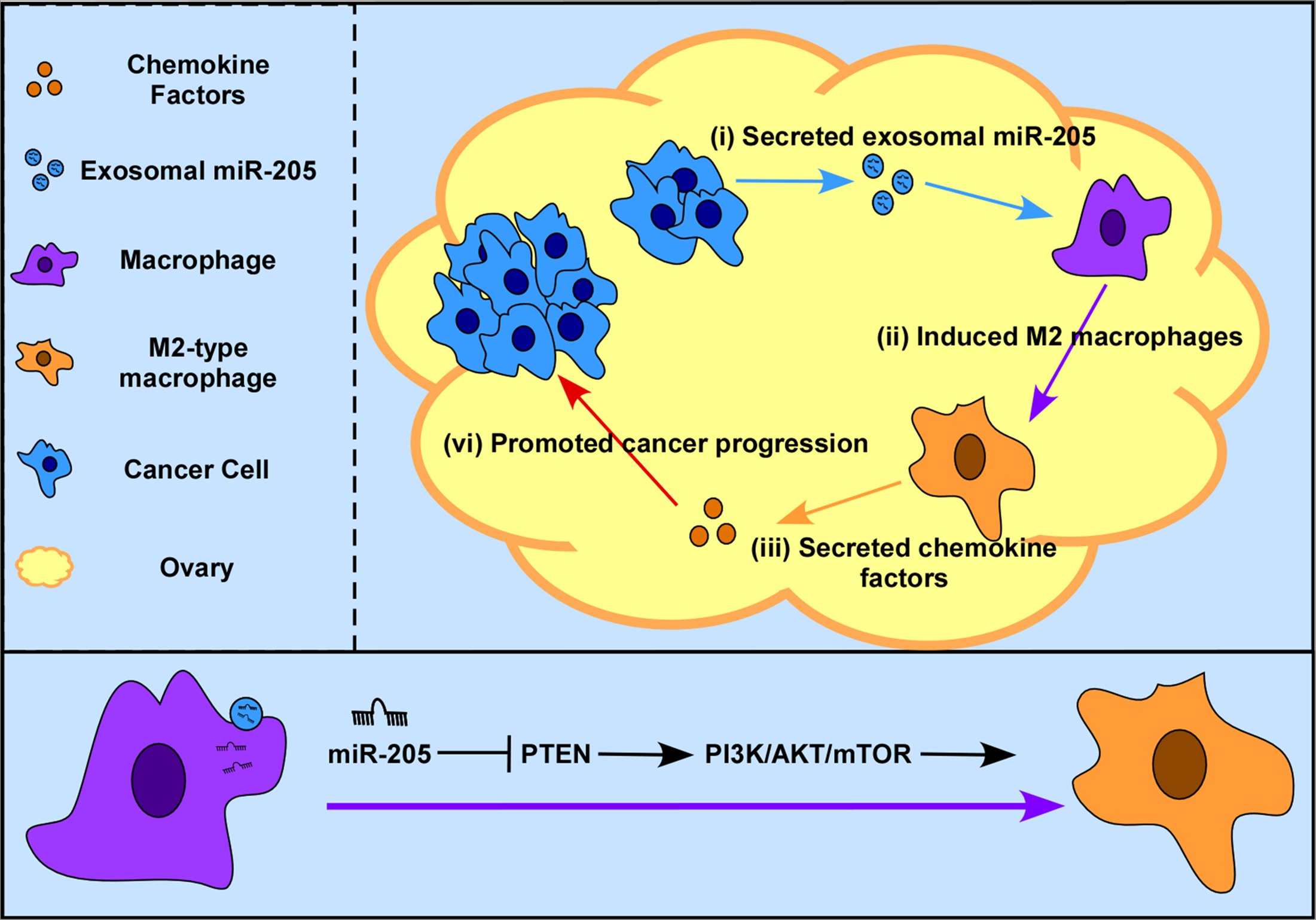{kind=link}