Plant material
The bark of an adult tree of Anadenanthera colubrina (Vell.) Brenan var. cebil (Griseb.) Altschul (Fabaceae) were collected in November 2018, during the dry season, in the city of Monte Alegre de Sergipe, Sergipe, Brazil (10° 2' 44” S and 37° 35' 4” W). The species was authenticated by phD Marcus Vinicius Meiado and deposited at the Federal University of Sergipe Herbarium (ASE nº 42361).
Preparation of plant extract
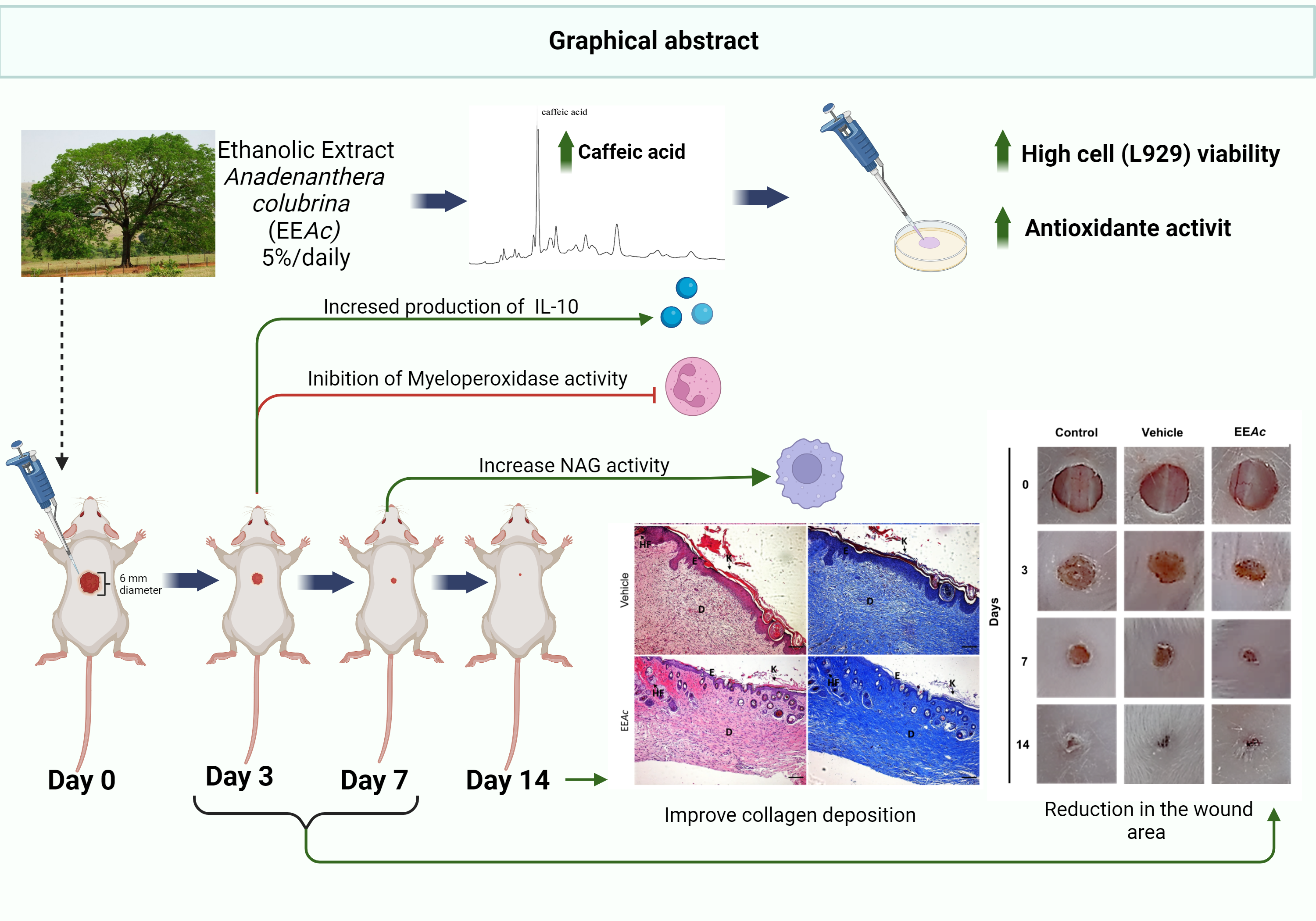
The bark was dried at 40°C (± 2) for 72 hours, ground, and weighed, yielding 1.1 kg of powder. This material underwent exhaustive maceration in absolute ethanol for 72 hours, a process repeated three consecutive times. The resulting liquid was filtered and subjected to rotary evaporation at 45°C (Fisatom®, São Paulo/SP, Brazil). The resulting dry extract (338 g) was stored at 2–8°C. For formulation purposes, 5 g of the extract were diluted in 100 mL of a vehicle composed of 2% Dimethylsulfoxide (DMSO; Merck, Brazil) in Propyleneglycol (PG),[22] resulting in the Ethanolic Extract of Anadenanthera colubrina (EEAc) at 5% [15, 20].
Characterization of EEAc by HPLC
The EEAc was diluted with methanol at a 5 mg/ml concentration. Both extract solutions were submitted to sonication for 30 minutes in an ultrasonic bath and were filtered on membrane filters (PTFE – 0.45 µm), before High-performance Liquid Chromatography coupled to Diode-Array Ultraviolet Detector (HPLC-DAD-UV) injection.
The HPLC-DAD-UV analyses were performed using a high-performance liquid chromatography system that consisted of a DGU-20A3 degasser, two LC-20AD pumps, an SIL-20A HT auto-injector, a CTO-20A column oven, an SPDM20Avp photodiode array detector (DAD) and a CBM-20A system controller (Shimadzu Co., Kyoto, Japan). The chromatographic separation was performed using a Shimadzu ODS analytical column 180 × 4.6 mm (5 µm particle size), equipped with a Shimadzu ODS guard cartridge system 20 x 4 mm (5 µm particle size). The mobile phase consisted of (A) ultrapure water with acetic acid (1.0%) and (B) methanol. The flow rate was 0.8 ml/min and the sample injection volume was 20 µL. The elution gradient started with 5% B (0.01–5 min); 5–15% B (5–12 min); 15–28% B (12–13 min); 28–28% B (12–13 min); 28–30% B (13–14 min); 30% B (14–22 min); 30– 31% B (23–25 min); 31–32% B (25–35 min); 32% B (35–39 min); 33–34% (40–45 min); 35–40% (50–52 min). Finalizing the analysis and returning to the initial conditions. The photodiode array detector was set at 280 nm for acquiring chromatograms. Compound identification was based on comparisons of absorption spectra and by co-injection with standard substances (based on retention times). Caffeic acid (C9H8O4), chlorogenic acid (C16H18O9), gallic acid (C7H6O5), ferulic acid (C10H10O4), myricetin (C15H10O8) and quercetin (C15H10O7) standards were obtained by Sigma-Aldrich® and prepared at a final concentration of 0.1 mg/mL with acetonitrile.
Quantification of total phenolics
The methodology for total phenolics content determination was based on the protocol proposed by Swain and Hillis (1959), with modifications adapted for a microplate format. A 12 µL aliquot of the extract was pipetted in quadruplicate into a 96-well plate. Subsequently, 12 µL of Folin-Ciocalteu reagent and 200 µL of distilled water were added to each well. After a 3-minute reaction time, 25 µL of saturated sodium carbonate solution (Na₂CO₃) was introduced. The plate was then kept at room temperature, and shielded from light. After one hour, the absorbance was measured using a spectrophotometer at 720 nm. Results were expressed as milligrams of gallic acid per gram of dry extract, determined using a standard curve with concentrations ranging from 25 to 200 µg/mL (y = 0.0092x + 0.0385; R² = 0.9976).
In vitro experiments
Antioxidant assays
In vitro tests were performed in triplicate, to determine the antioxidant capacity of EEAc. The extract concentrations used were 25, 50, 100, and 200 µg/mL, and the concentration required to inhibit half of the maximum evaluated parameter (IC50) was calculated. Trolox (Sigma Aldrich, USA) was used as a standard of the antioxidant. The antioxidant capacity against the 2,2-diphenyl-1-picrylhydrazyl radical (DPPH; Sigma-Aldrich, USA) was measured by adding 50 µL of extract to 150 µL of DPPH at a concentration of 400 µM/L in a microplate. After 30 minutes of rest, protected from light, the reduction of the DPPH radical was measured at 515 nm using a plate reader (Biotek, Synergy Mx).
The ABTS radical assay [2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)] was performed adding 30 µL of the sample in a microplate and was added to 300 µL of the ABTS radical solution (1,25 mL of a 7 mM ABTS solution in 22 µL of a 140 mM potassium persulfate (K₂S₂O₈) solution). For these tests, the results were expressed as a percentage of inhibition [% inhibition = [(control − test)/control] × 100], based on the absorbance values, from which the IC50 values were calculated.
The ferric-reducing antioxidant power (FRAP) assay was conducted in a dark environment by adding 9 µl of each extract concentration in triplicate to a microplate., followed by 27 µl of distilled water, and 270 µl of the FRAP reagent, consisting of TPTZ, ferric chloride solution, and acetate buffer (0.3 M, pH 3.6) in a 1:1:10 ratio. The microplate was then incubated at 37°C for 30 minutes. After incubation, absorbance was measured using a spectrophotometer at 595 nm. Results were expressed in µM Trolox equivalents, calculated using the equation y = 0.0008x + 0.102.
Cytotoxicity in fibroblasts
Cell viability of the fibroblast cell line, L929, was assessed using the methylthiazolyl tetrazolium bromide (MTT) colorimetric assay. Cells were cultured in DMEM (Sigma Aldrich, USA) containing 10% fetal bovine serum (Life Technologies, India), 1% penicillin (Life Technologies, India)/streptomycin (Life Technologies, India), and maintained in an atmosphere of 5% CO2 at 37°C. Fibroblasts were seeded in 96-well culture plates (2 × 104 cells/well) and treated with EEAc at concentrations of 12.5, 25, 50, 100, and 200 µg/mL for 24 hours. Then, MTT (0.5 mg/mL in phosphate-buffered saline (PBS)) was added to the cells and incubated at 37°C for 3 hours. After MTT removal, DMSO was added to solubilize the formazan crystals, and the absorbance was measured at 570 nm. The tests were conducted in triplicate and normalized by considering the control absorbance as 100% viability.
Wound healing in vivo
Swiss albino mice (Mus musculus), female, nulliparous, healthy, weighing between 25–30, aged 8–12 weeks. Were kept individually in polypropylene cages, with 21 ± 2°C temperature, 60 ± 5% humidity, and a 12-hour light / dark cycle, in addition to food and water ad libitum.
Excision of skin wounds and topical treatments
Before the induction of the lesions, the animals underwent a seven-day acclimatization period. Ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) were used as anesthetic medications. Following anesthesia, the dorsothoracic region was shaved, and antisepsis was performed using 0.5% alcoholic chlorhexidine solution. Circular cutaneous excision was performed with a 6 mm metallic punch [23]. The animals were randomly assigned to three groups: control (treated with sterile saline 0.9%), vehicle (2% DMSO in PG), or EEAc. Treatments were administered immediately after wound induction and repeated every 24 hours for 14 days. Each wound received 30 µL of the respective treatment (equivalent to 1.5 mg of EEAc per wound per day).
Wound contraction
Wounds were measured on days 0 (initial wound), 3, 7, and 14 using a digital caliper (n = 5–7/group). The wound area was calculated according to the equation: Wound area (mm²) = π.R.r, where π = 3.1416; R = larger radius and r = smaller radius. Wound closure was determined using the equation: Wound closure (%) = Wi-Wd/Wi x 100, where Wi equals the initial wound area and Wd the wound area on the measured days [24]. After obtaining this parameter, the % wound area remaining open was calculated by reducing the % closure value by 100.
Tissue collection
The animals were euthanized using an overdose of ketamine (300 mg/kg, i.p.) and xylazine (30 mg/kg, i.p.). Skin fragments, including both the intact area and the wound site, were collected using an 8 mm punch. The collection was performed at different time points depending on the healing progression, including days 1, 3, 7, and/or 14 post-injury.
Histopathological analysis
Animals designated for wound closure analysis (n = 5/group) underwent histological evaluation on the 14th day post-injury. Tissues were sectioned in half into two fragments, cut in a microtome (5 µm), and stained with Hematoxylin & Eosin (H&E) or Masson's Trichrome (Masson’s). Subsequently, the tissue sections were examined (10 different areas) under an optical microscope (Nikon, Japan), and images were captured using a digital camera (Coolpix 4500, Roper Scientific, Japan). Analysis was conducted by two researchers independently, in a blinded manner. A scale ranging from 0 to 3 was utilized to assess various parameters including leukocyte infiltrate, vascularization, re-epithelialization, and collagen deposition. The scoring system was as follows: 0 for absent, 1 for mild, 2 for moderate, and 3 for accentuated [25].
Inflammatory essays
Myeloperoxidase and N-acetyl-β-D-glycosaminidase activity
Quantification of myeloperoxidase (MPO) activity was performed for indirect assessment of neutrophil infiltrate on days 1, 3, and 7 of healing (n = 5–7/group/time). The tissues were homogenized in PBS and Hexadecyltrimethylammonium Bromide 0.5% (HTAB 0.5%), in the proportion of 1mL/100mg of tissue. Following homogenization, the samples were centrifuged for 2 minutes at 14000 rpm. A volume of 20 µL of supernatant was used for the reaction with 200 µL of O-dianisidine solution and, finally, the absorbance of the samples was determined at 460 nm [26].
For the indirect determination of the macrophage infiltrate in the lesions, N-acetyl-β-D-glycosaminidase (NAG) activity was quantified on days 1, 3, and 7 of the repair process (n = 5–7/group/time). Tissue samples were homogenized in cytokine extraction buffer (PBS containing protease inhibitor cocktail, Phenylmethylsulfonyl fluoride (PMSF), Sodium chloride (NaCl), Ethylenediamine Tetra-acetic acid (EDTA) and Tween 20) and centrifuged. The pellet was resuspended in Buffer containing 0.2% NaCl and 1.6% NaCl with 5% glucose, posteriorly. Subsequent steps involved homogenization, centrifugation, and resuspension of the pellet in NaCl 0.9% and Triton x-100 0.1% (1:1). The supernatant was mixed with 0.767 mg/ml of p-nitrophenyl-N-acetyl-β-D-glycosaminide for the NAG assay. The plate was incubated at 37ºC for 30 minutes and the reaction was stopped with buffer containing 0.2 M glycine. The samples were analyzed at 405 nm. Finally, the result was normalized by the weight of the tissue and expressed in Optical Density (OD)/ 100 mg of the tissue [27].
Quantification of cytokines
The tissue samples on days 1 and 3 of repair (n = 5–7/group/time) were subjected to quantification of cytokines TNF-α (Tumor Necrosis Factor–α) and IL-10 (Interleukin 10) using ELISA kits according to the manufacturer's protocol (Peprotech®, USA).
Statistical analysis
The data were submitted to the Shapiro-Wilk normality test. According to the data normality, a one-way or two-way ANOVA test was performed with Bonferroni post-test, or the Kruskal-Wallis test was used with Dunn's post-test. Data were expressed as mean ± SEM or median and interquartile range for scores of histological parameters. Statistical analysis was performed using the GraphPad Prisms® 8.0 software, with p < 0.05 being considered significant.
{kind=link}