Photophysical properties and theoretical analysis. A series of PAHs with buckybowl-based structure was prepared in accordance with our prior research38–41 and denoted TP, TPy, TH, and TQ (Fig. 1a). All the PAHs demonstrate visible-light absorption (1×10− 5 M in THF, Table S1), indicating their ability to perform photochemical reactions under natural light. The absorption and fluorescence characteristics of these PAHs exhibit a bathochromic shift from TP to TQ, which is attributed to the extension of π-conjugation (Fig. 1b, Table S2).42. This indicates the tunable energy level of the designed phosphors. Delayed emission spectra were recorded in 2-MeTHF (1 × 10− 5 M) at 77 K (Fig. 1c). Notably, TP, TPy, and TH display phosphorescence emission at 77 K, while TQ shows no phosphorescence emission. Thus, TQ is not capable of generating triplet excitons. In this context, assessing the triplet exciton yield of PAHs is pivotal because this parameter is related to the sensitization efficiency of photosensitizers.
Theoretical calculations facilitate an understanding the photophysical properties of the PAHs. Highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of PAHs are distributed throughout the molecule, indicating no obvious intermolecular charge transfer (Figure S1). With the increase of conjugation, the energy gap (Eg) between HOMO and LUMO decreases continuously, which is consistent with the UV-vis absorption spectra (Fig. 1b). To gain further insight into electronic transition process of the molecule, the excited state PAHs were calculated based on time-dependent density functional theory (TD-DFT). The hole-electron population of each state was evaluated using Multiwfn 3.8 package.43
The evaluation of the ISC ability of a molecule can be conducted through a joint analysis of the spin-orbit coupling (SOC) values and the energy difference (ΔEST) between single-triplet states.44–46 The lowest singlet state (S1) and the lowest triplet state (T1) of PAHs have the same orbital configuration with a large proportion, suggesting an efficient ISC channel (Table S3-6). The SOC constants < S1|HSO|T1 > of TP, TPy, TH, and TQ are 0.16, 1.38, 0.71, and 0.74, respectively. Additionally, the electron-hole distribution of S1 and T1 of PAHs is highly similar (Figure S2), which further ensures that the singlet excitons are easily spin-flipped into triplet excitons. TP, TPy, and TH exhibit small ΔEST values of 0.15 eV, 0.20 eV, and 0.17 eV, respectively (Fig. 1d), implying that the presence of additional ISC channels. In contrast, TQ demonstrates a large energy difference (0.78 eV), significantly impeding the ISC process, despite the comparable S1 and T1 electron-hole distributions in TQ molecules. Thus, TQ do not emit phosphorescence, which aligns with the delayed spectrum observed for TQ at 77 K. Consequently, TPy exhibits the highest SOC constants and generates the most triplet excitons, suggesting that TPy possesses the best photosensitizing ability among the four PAHs.
PAHs photosensitizers for photocatalytic CO 2 reduction. To reveal the effects of the photophysical processes in the four PAHs on CO2 photoreduction, photocatalytic activities of these molecules were investigated in 5 mL CO2-saturated CH3CN/H2O (v/v = 4/1) solution containing [Fe(qpy)(OH2)2]2+ catalyst (referred to as CAT) and electron donors (EDs): triethylamine (TEA) and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH).47,48, TEA is used as a deprotonated Brønsted base to ensure the irreversibility of the more potent sacrificial BIH.49 The TONCAT of TPy-containing system is as high as 896 (22.41 µmol CO), followed by TH, while the TP and TQ molecules are almost inactive (Fig. 2a). These results reveal that the efficient ISC processes of TPy results in active photosensitizing properties for CO2 photoreduction. Importantly, the organic fused-ring molecule with the high structural stability and a large π-conjugation for charge delocalization38 after the photoredox showed great advantage for CO2 photoreduction.
Further, nanosecond transient absorption spectra of kinetic decay were employed to understand the photosensitizing process of the organic phosphors in CO2 photoreduction system. The TP, TPy, and TH molecules show long-lived triplet excitons capable of sufficient intermolecular electron transfer between the photosensitizers and photoreaction substances, with triplet exciton lifetimes of 74.56 µs, 48.74 µs and 102.58 µs, respectively (Fig. 2b). These findings establish the close relationship between the capacity of PAHs to generate triplet excitons and photosensitization efficiency, particularly in the presence of long-lived triplet excitons. Moreover, the stability of the triplet excitons is a vital factor in ensuring sufficient time for intermolecular electron transfer to boost photosensitization. TPy with the highest ISC efficiency also leads to a relatively short triplet excited state lifetime, potentially impairing the sensitization ability. In addition, TPy shows poor solubility in the photocatalysis solution and gradually aggregates (Figure S3), leading to a decline in photocatalytic efficiency. Therefore, stabilizing both TPy molecules in solution and its triplet excitons may further promote photosensitization efficiency.
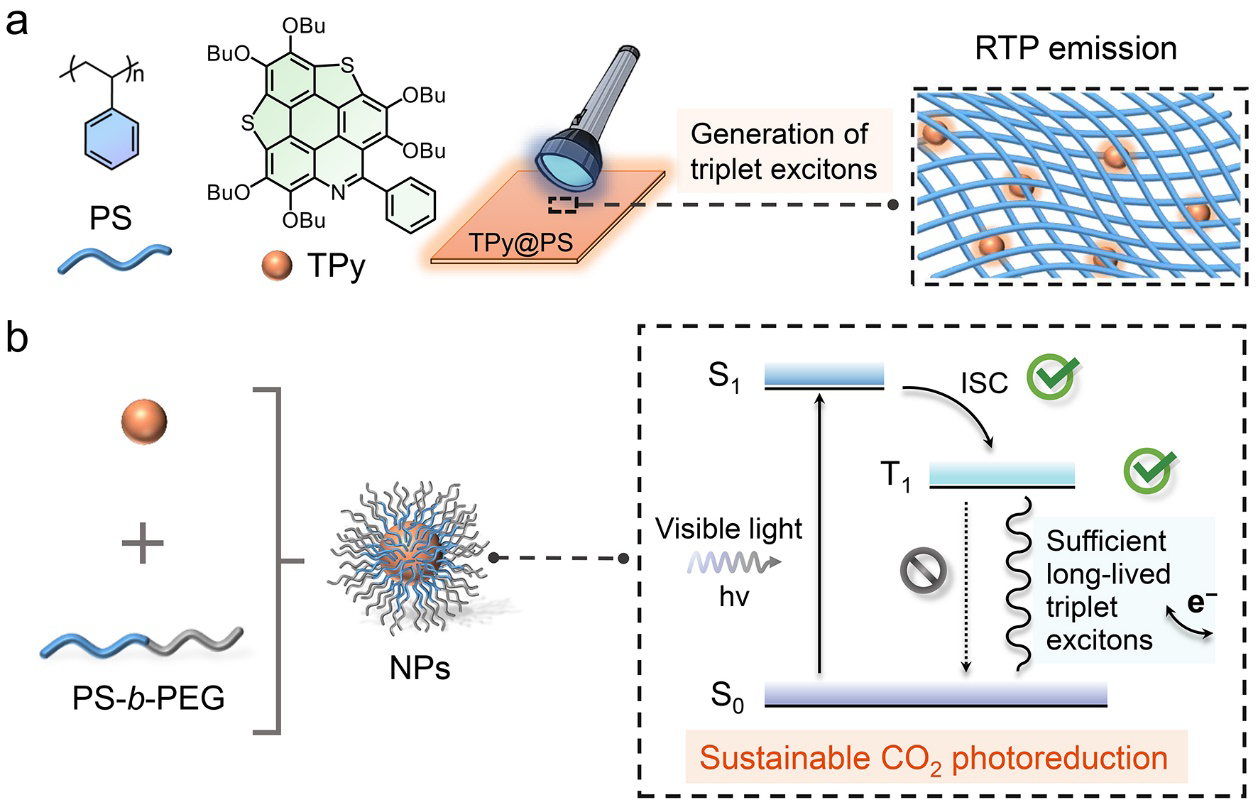
RTP phenomenone of the doped system. A polymer host can effectively inhibit molecular motion and stabilize triplet excitons.24,25,50–52 Herein, inspired by the preparation of organic RTP materials in our previous work,51 PS was chosen as a polymer matrix, and a series of TPy@PS doped films with different mass ratios were prepared. In prompt spectra, the highest luminescence peak is observed at a mass ratio of 1:75 (Figure S4), attributed to the phosphorescence peak of molecularly dispersed TPy at 620 nm (Fig. 1c). Of note, TPy molecule shows no phosphorescence at room temperature (Figure S5). This indicates that PS is capable of acting as a rigid matrix to inhibit the non-radiative transition of TPy, thereby stabilizing the triplet excitons and facilitating RTP. The robust interaction between the phenyl ring in PS and PAHs can avoid strong π-π stacking interaction between photosensitizers, enhancing triplet exciton stability.31 Therefore, a guest@host doping mass ratio of 1:75 was used for subsequent experiments. In vacuum delayed spectra, TPy@PS shows a strong phosphorescence emission (Fig. 2c, Figure S6), with a phosphorescence quantum yield (QY) is up to 14.84% (Fig. 2d). In comparison, TP@PS and TH@PS have very weak phosphorescence (Figure S7), which further indicates their weak triplet exciton generation ability. TPy@PS has the highest phosphorescence efficiency, followed by TH@PS and TP@PS (Fig. 2d), which is consistent with theoretical calculations of PAHs (Fig. 1d). These results suggest that utilizing PS as a polymer matrix can stabilize the triplet excitons, leading to improved RTP emission. Importantly, this straightforward doping strategy enables the rapid screening and evaluation of RTP materials as efficient photosensitizers using spectroscopy.
Photophysical properties of self-assembled NPs. To further utilize the triplet excitons stabilized by the PS matrix for CO2 photoreduction, TPy and the amphiphilic block copolymer PS-b-PEG were assembled via microfluidic technology (Fig. 3a).35,53 The properties of NPs are highly related to their size and morphology.17,54,55 Large NPs may prevent the transfer of triplet excitons to the reaction substance, while triplet excitons may not be stable enough in small NPs.35 It is vital for the preparation of monodisperse NPs with controllable aggregates and high uniformity by microfluidic technology. During the self-assembly process, the hydrophobic PS segment within PS-b-PEG acts as the host, wrapping TPy molecules and stabilizing the triplet excitons. Meanwhile, the hydrophilic PEG segment enables the stable dispersion of the NPs in water. TPy do not emit phosphorescence in either oxygenated or deoxygenated water (Fig. 3b). Notably, the NPs also do not emit phosphorescence in oxygenated water, but emit in argon-deoxygenated water. This phenomenon shows that the hydrophobic PS segment is capable of stabilizing the triplet excitons without hindering the interaction of TPy with various components in the solution, thus manipulating energy decay of triplet excitons. Consequently, the capacity of NPs to generate triplet excitons is evaluated by their phosphorescence emission intensity in deoxygenated water.
Modifying the hydrophilic-hydrophobic ratio of the amphiphilic polymer can tune the particle size of the self-assembled NPs.55,56 Therefore, a series of diblock polymer was prepared with different PS15k-b-PEGxk hydrophilic-hydrophobic ratio (x = 2, 5, 10, 23, 35). With increasing hydrophilic ratio, the size of the NPs becomes smaller (Figure S8). However, when x = 2, insufficient hydrophilic segments result in the formation of unstable NPs that are prone to agglomeration and precipitation in the solution and during the catalytic process. Changing the hydrophilic PEG segment from 2k to 23k in deoxygenated water leads to a significant enhancement of phosphorescence intensity and a prolonged phosphorescence lifetime (Fig. 3b, Figure S9). However, an excessive hydrophilic ratio diminishes the hydrophobicity of the PS segment, causing loose wrapping and a subsequent decrease in phosphorescence intensity and lifetime. This trend is further validated by total and phosphorescence QYs, confirming that NPs-23k generate the highest triplet excitons (Fig. 3c). In addition, the the UV-vis absorption test shows that the structural stability of NPs-23k is significantly higher than that of TPy under long-term irradiation (Figure S10), which clearly indicates the protection and stability of PS for the TPy structure. Finally, the hydrodynamic diameter of NPs-23k (~ 160 nm, polydispersity index: 0.19) is determined by dynamic light scattering (DLS). The insert transmission electron microscope (TEM) image shows the spherical morphology of NPs-23k (~ 110 nm) in dry state (Fig. 3d, Figure S11). These experiments confirm that NPs-23k exhibit excellent photobleaching resistance, uniform particle size, good dispersion, large surface area, and the highest yield of triplet excitons. Consequently, it can be reasonably inferred that NPs-23k may exhibit a superior sensitization effect on the reactants in the catalytic system.
Self-assembled NPs for photocatalytic CO 2 reduction. Finally, the performance of NPs-23k for photocatalytic CO2 reduction was investigated. NPs-23k generates long-lived triplet excitons with a lifetime of 96.19 µs, which is twice of TPy molecule (Fig. 2b, 4d). Under the same conditions, the catalytic efficiency of NPs-23k-containing system is increased by nearly 50% compared to that of TPy, the CO yield can reach 33.40 µmol with a TONCAT value of 1336 (Figure S12). However, the catalytic effect of the system without a filter is inferior. The CO yield of the NPs-23k-containing system is only 7.72 µmol (Figure S13), a value comparable to that observed under irradiation with a 395 nm monochromatic light source (Figure S14). This observation was attributed to the disruption of the organic structure caused by prolonged exposure to high-energy ultraviolet radiation. The above findings demonstrates that utilizing PS-b-PEG with host function effectively stabilizes the triplet excitons of TPy. Moreover, after self-assembly, spherical NPs-23k are uniformly dispersed in the catalytic system (Figure S15), which substantially increases the surface area for electron transfer to the surrounding medium. Additionally, in contrast to visible TPy aggregates (Figure S3), monodisperse NPs-23k can reduce exciton losses caused by triplet exciton annihilation in aggregates57, thus improving the sensitization efficiency. Next, the concentrations of the photosensitizer and EDs in the photocatalytic system were optimized (Table 1). The quantification of the gaseous products shows that the C/H selectivity of photoreduction CO2 to CO is about 95% under all conditions. The TONCAT of NPs-23k-containing system reaches 2041 (51.02 µmol CO) within 18 h illumination (100 mw∙cm− 2, λ > 400 nm) under the optimal conditions (Table 1, Group 2). Clearly, the simple self-assembly method employed in this work endows TPy with more stable and longer-lived triplet excitons. This enables the facile transfer of electrons to surrounding compounds, thus favoring the photosensitization process.
Table 1
The results of photocatalytic reduction of CO2 to CO.[a]
| Group | Photo-sensitizers (µM) | BIH (mM) + TEA (mM) | CO (µmol) | H2 (µmol) | TONCO | TOFCO[b] (h− 1) | SelCO (%) |
| 1 | NPs-23k | 10 | 18 + 0.3 | 26.50 | 1.51 | 1060 | 132.7 | 94.6 |
| 2 | NPs-23k | 20 | 18 + 0.3 | 51.02 | 2.77 | 2041 | 255.5 | 94.9 |
| 3 | NPs-23k | 20 | 36 + 0.6 | 33.13 | 1.94 | 1325 | 166.0 | 94.5 |
| 4 | TPy | 10 | 18 + 0.3 | 17.54 | 0.91 | 702 | 89.2 | 95.1 |
| 5 | TPy | 20 | 18 + 0.3 | 33.77 | 1.63 | 1351 | 171.7 | 95.4 |
| [a]In 5 mL CO2-saturated CH3CN/H2O (v/v = 4/1) solution with 5 µmol CAT under irradiation with a 300 W Xenon lamp (100 mw·cm− 2, λ > 400 nm, irradiation time: 18 h). [b]6 h |
To investigate the stability of catalytic system under irradiation, recycle experiments were performed by re-adding NPs-23k, CAT, and EDs after 6 h irradiation (Figure S16). Following the addition of CAT and EDs in the 2nd cycle, the photocatalytic activity of the reaction system is almost entirely restored. This indicates that the inactivation of the photocatalytic system is primarily caused by the decomposition or consumption of CAT and EDs. To assess the stability of TPy and NPs-23k, consumed CAT and EDs were re-added after each 6 h cycle. As shown in Fig. 4a, after three cycles (18 h), the sensitization ability of NPs-23k remains at 93.5%, while that of TPy is only 74.9%. Remarkably, the total CO yield of the NPs-23k-containing system reaches 113.6 µmol. This indicates that the stability of NPs-23k photosensitizers is higher than that of TPy, because the vibrational relaxation of TPy is not suppressed in the absence of the PS coating, which leads to declining stability under long-term irradiation. These results further emphasize the significance of assembling TPy with PS-b-PEG. The self-assembly strategy prolongs the triplet excitons lifetime, enlarges the area of electron transfer with the medium, prevents the loss of triplet excitons, thus effectively improving the photosensitization cyclic ability of the catalytic system.
Mechanism analysis of photocatalytic system. In the absence of photosensitizer (TPy/NPs-23k), CAT, EDs, light, or CO2, trace or no CO was detected (Table S7), indicating all of these factors are indispensable for CO2 photoreduction. Moreover, to confirm the source of CO, isotope-labeling experiments with 13CO2 as the carbon source were performed, and 13CO was detected as the main product (Figure S17). These results prove that the CO product is indeed generated from CO2 instead of the decomposition of organic photosensitizers. To elucidate the photocatalytic mechanism, the thermodynamic feasibility of intermolecular electron transfer was evaluated, and cyclic voltammetry (CV) was conducted to study the key components of the reaction system (TPy, CAT, and BIH) in a degassed acetonitrile solution (Figure S18, Table S8). The oxidation potential of BIH is far more negative than the excited state reduction potential of TPy (*Red) (0.52 V < < 1.95 V vs. SCE), and the excited state oxidation potential of TPy (*Ox) is more negative than the reduction potential of CAT (-1.21 V < -1.05 V vs. SCE) (Fig. 4b). The results indicate that both the reduction and oxidation quenching mechanisms are thermodynamically feasible in the studied photocatalytic system. However, the potential difference for the reduction quenching mechanism is greater than that for the oxidation mechanism (-1.43 V vs. -0.16 V). In addition, the absorption spectra of TPy remained unchanged before and after adding the BIH or CAT (Figure S19), revealing the absence of intermolecular electronic interaction between TPy and CAT or BIH under ground state. Phosphorescence quenching experiments were performed with NPs-23k using CAT or BIH as quenchers. At the concentration scale of CAT and BIH in the photocatalytic system, the phosphorescence intensity of NPs-23k is more efficiently quenched by BIH compared to CAT (Fig. 4c). These findings suggest that the photocatalytic process is primarily governed by the reduction mechanism, which was further supported by nanosecond transient absorption studies (Figure S20, Fig. 4d and 4e).
Nanosecond transient absorption spectra were utilized to probe the excited state properties and intermolecular electron transfer of NPs-23k and TPy. As shown in Figure S20a and b, a strong bleaching peak below 350 nm is observed upon pulsed laser excitation, while positive peaks at 360 nm and beyond 450 nm correspond to the excited states of NPs-23k and TPy.6,22 The excited state map of NPs-23k is similar to that of TPy, indicating that the excited state type of TPy remains unchanged after self-assembly. When BIH is added to the NPs-23k solution, the peak at 360 nm shows a lower intensity and narrower peak width (Figure S20b and c). In addition, the positive signal above 530 nm vanishes and a new short negative peak appears at 410 nm. This new peak is assigned to the reduced NPs-23k with a long lifetime of 191.48 µs (Figure S20c, Fig. 4e). The subsequent introduction of CAT reduces the lifetime of NPs-23k to 13.27 µs, indicating rapid electron transfer from the reduced NPs-23k to CAT, and the excited state map of the entire system is restored (Fig. 4e, Figure S20b and d). Analogous observations are made in TPy-containing systems (Figure S21 and 22). Consequently, the electron transfer route of the NPs-23k-containing and TPy-containing photocatalytic systems are all classified as reduction mechanism (Fig. 4f).
{kind=link}