Cells
Anti-CD362+-selected hUC-MSC were cultured from ethically-sourced human umbilical cord tissue obtained from Tissue solutions Ltd. (Glasgow, U.K). Primary isolation and expansion cultures of CD362+ hUC-MSC was carried out as previously described (19, 23, 27). Cryopreserved vials (1x107 in 1mL) of anti-CD362+-selected hUC-MSC were thawed, transferred into 9mL of phosphate buffered saline (PBS). After live/dead analysis either via trypan blue dye or automated cell counter (NucleoCounter® NC-200™, Chemometec A/S, Denmark), the required numbers of cells were pelleted via centrifugation at 400xg for 5 minutes and the cells were re-suspended in 100µl of sterile saline for intra-peritoneal (i.p.) or i.v. injection.
Animal Procedures
All animal procedures were carried out under the license (no. 255/17) from the Animal Experimentation Ethical Committee, University of Barcelona and under authorisation (AE19125/P082 and AE19125/P066) from the Health Products Regulatory Authority, Ireland, and approved by the NUI Galway Animal Care Research Ethics Committee. All procedures were performed in licensed animal facilities at NUI Galway and University of Barcelona.
For the LPS model of endotoxemia, 10–12 week-old male C57BL/6 mice from Charles River Ltd., Kent, UK were used. Mice were injected i.p. with 5µg/g LPS (LPS 0111:B4, catalogue no. L2630, Sigma Aldrich, UK) in 100µl of sterile saline. The mice were housed in groups of 3–5 mice/cage during the study in individually ventilated cages. Single i.p. injections of 2.5 x 105 hUC-MSC or equal volumes of vehicle (sterile saline) were administered 4 hours after LPS injections. The animals were monitored every 4 hours until the end of the study using a distress score sheet and support measures according to a pre-determined protocol. Humane euthanasia was performed at the defined experimental end-point or earlier if animals exceeded the pre-defined severity score threshold. At the time of euthanasia, peritoneal exudates were collected for flow cytometry analysis by carefully flushing 5mL of sterile PBS into and out of the peritoneal cavity.
Cecal ligation and puncture (CLP) was performed on 8–12 week-old, male C57BL/6 mice (Charles River Ltd., UK). The mice received buprenorphine 0.1 mg/kg (Richter Pharma AG, Austria) subcutaneously 25–30 minutes before the procedure and were anesthetized with 1.8-2% isoflurane (with O2 flow of 0.5L/min) at NUI Galway or with Anesketin (100mg/mL; Dechra Veterinary Products SLU, Spain) and Rampun (20 mg/mL; Bayer, Germany) at University of Barcelona. The lower half of the abdomen was shaved and cleaned with 4% chlorhexidine or povidone-Iodine and incised 1cm vertically along the midline. The cecum was externalized and the distal 50% was ligated using 4.0-6.0M sutures. Cecal material was released by ‘through and through’ puncture with a 21-gauge needle and a drop of fecal matter was exuded before reinstating the cecum into the peritoneal cavity and suturing the muscle and skin closed. Sham-operated mice underwent an identical procedure, including opening the peritoneum and exposing the bowel, but without ligation and perforation of the cecum. Mice received 0.5mL of Gelofusine (Braun Melsungen AG, Germany) by i.p. instillation prior to wound closure. Post-operative support consisted of buprenorphine diluted given subcutaneously (s.c.) every 8–12 hours until the pre-determined end-points (48 or 72 hours for individual experiments). Administration of 1x106 hUC-MSC or equivalent volumes of vehicle (sterile saline) was carried out i.v. via the tail vein at 4 hours or at 4 and 28 hours following CLP. Frequent monitoring and support was carried out according to an ethically-approved protocol. Humane euthanasia was performed at the defined experimental end-point or earlier if animals exceeded a pre-defined severity score threshold.
Blood Sampling and Tissue Procurement
Venous blood samples to a maximum volume of 20µL were drawn intermittently from tail and facial veins by aseptic technique using 25 − 21 gauge needles and were collected into heparin (VWR International, Dublin, Ireland)-containing tubes. A terminal blood sample was drawn by cardiac puncture at the time of euthanasia. Serum was collected in micro-tubes with serum gel and clotting activator (Sarstedt, Wexford, Ireland). Plasma and serum samples were prepared by centrifugation at 10,000xg for 10 minutes. Serum samples were stored at -80°C and subsequently analysed for biochemical parameters by NationWide Laboratories (Lancashire, UK). Spleen, lungs, kidneys and liver were dissected immediately after euthanasia.
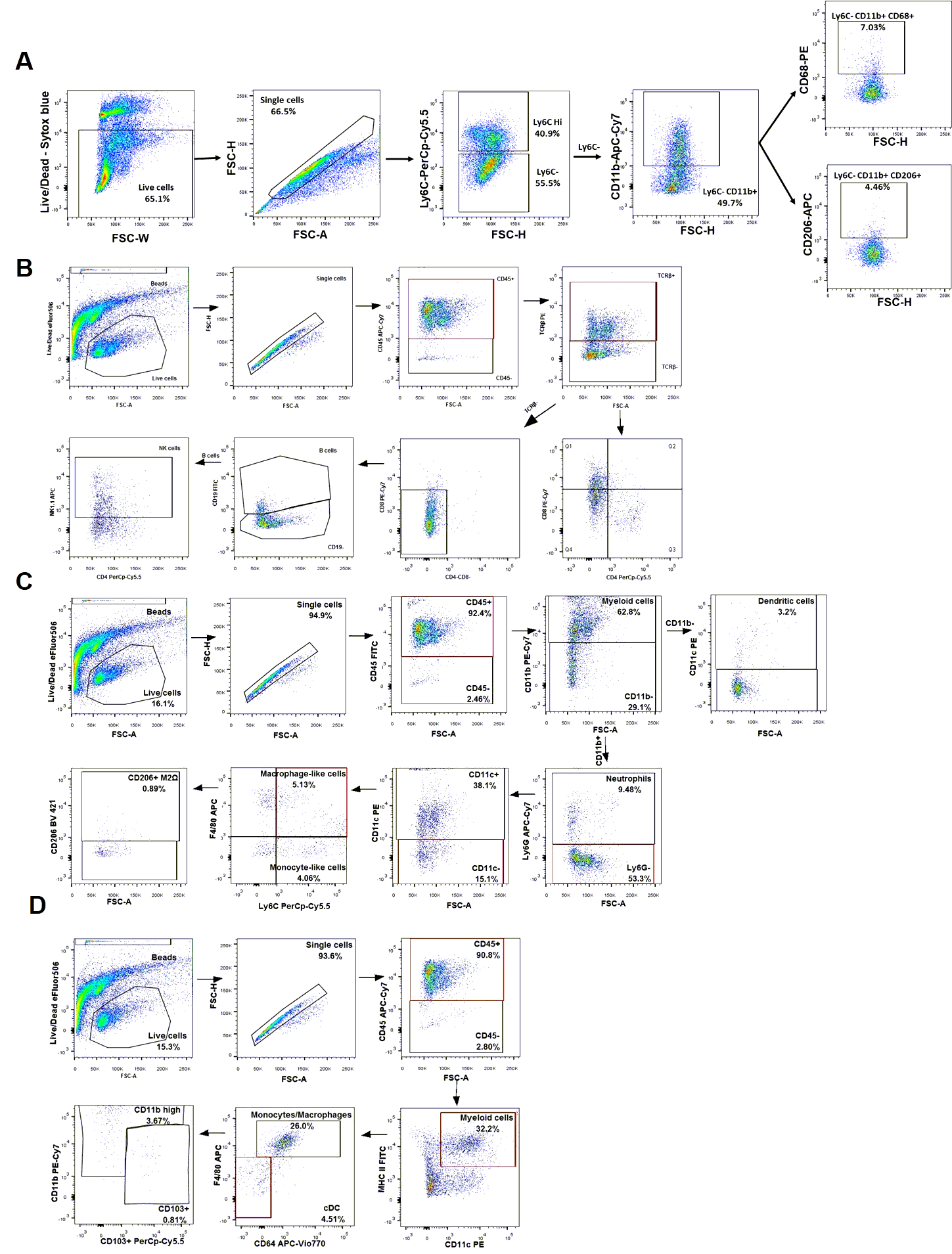
Multicolour flow cytometry: For flow cytometry of mouse peritoneal macrophages (LPS model), 100µL of freshly-prepared peritoneal exudate cells re-suspended in FACS buffer (PBS, 2% fetal calf serum and 0.05% sodium azide) were labelled with the following combinations of fluorochrome-labelled antibodies at 4°C for 30 minutes: anti-Ly6C -PerCP-Cy™5.5 (clone-AL-21), anti-CD11b -APC-Cy7 (clone-M1/70), anti-CD68-PE (F1/11), anti-CD206-APC (MR5D3) from BD Pharmingen™ (BD Bioscience, Berkshire, UK). Cell viability was analysed by SYTOX™ blue dead cell stain (Thermo-Fisher Scientific, Dublin, Ireland) according to manufacturer’s instructions. For flow cytometry of kidney cells, single-cell suspensions were prepared from freshly-dissected kidneys by collagenase/DNase digestion and were enriched for CD45+ bone marrow-derived cells by magnetic column separation. Kidney cell suspensions were stained with panels of fluorochrome-coupled monoclonal antibodies. A detailed protocol for preparation and flow cytometry analysis of kidney cell suspensions is available in Supplementary Methods. For all flow cytometry analyses, labelled cells were washed and re-suspended in FACS buffer and immediately analysed on a FACS Canto II cytometer (BD Biosciences). Data files were subsequently analyzed using FlowJo v6 software (Ashland, OR, USA). Details and examples of the gating strategies used to define and enumerate specific immune cell subpopulations are provided in Supplementary Table 1 and Supplementary Fig. 1.
Immunoassays
Plasma neutrophil gelatinase-associated lipocalin (NGAL) concentration was quantified with the mouse Lipocalin-2/NGAL Duo-Set ELISA Development kit (R&D Systems, Minneapolis, MI, USA) according to the manufacturer’s suggested protocol (details in Supplementary Methods). For multiplex quantification of cytokines and chemokines in serum, the Bio-Plex Pro mouse cytokine standard 23-plex assay (Bio-Rad, Accuscience) was used according the manufacturer’s instructions. Samples were analysed on a Bioplex 200 multiplex ELISA system (Bio-Rad, Accuscience).
Histology and Immunohistochemistry
Kidneys were dissected and placed in 10% neutral buffered formalin for 24 hours at room temperature before being processed in a Leica Tissue Processor ASP300. Tissues were wax-embedded in a Leica EG1150 wax embedder fitted with EG1130 Cold Plate, and 5-µm sections were cut using a Leica RM12235 microtome. Sections were transferred to Superfrost Plus microscope slides (Fisher Scientific Ireland) and dried overnight at room temperature. Histologic staining of sections for hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) were performed using standard protocols (details provided in Supplementary Methods).
For immunohistochemical staining, kidney tissue sections were dewaxed in xylene then hydrated in graded ethanol solutions. Heat-mediated antigen retrieval was performed with Tris/EDTA buffer pH 9.0 at 90°C for 30 minutes. Sections were treated with 0.3% H2O2, and incubated with avidin-biotin blocking solution (SP-2001, Vector Laboratories, Burlingame, CA, USA) to reduce nonspecific staining. Next, the slides were incubated for one hour at 4°C with rabbit anti-mouse NGAL monoclonal antibody (1:2000; ab216462, Abcam, Cambridge, UK), followed by incubation with biotinylated goat anti-rabbit IgG secondary antibody (BA-1000). For colorization, an avidin-biotin horseradish peroxidase complex (Vector Laboratories) and 3,3-diaminobenzidine substrate solution (Sigma-Aldrich) were applied to the slides at room temperature for 30 and 5 minutes respectively and the slides were counterstained with Gill no. 3 hematoxylin (Sigma-Aldrich) for 30 seconds. Negative control slides were prepared by staining under identical conditions but without adding the primary antibody.
Semi-quantitative Scoring of Kidney Tissue Sections
Stained sections of kidney were analyzed in blinded fashion by light microscopy at 40X magnification using an Olympus BX43 bright-field microscope (Olympus, Center Valley, PA) and with IS TCapture software (Tucsen Photonics Co., Fujian, China). For each kidney, twenty non-overlapping fields of a stained section were captured, and the positively stained area was scored by a blinded observer for (A) tubular dilatation, cast and necrosis (PAS) and (B) NGAL expression (28) Scoring was carried out on a 0–4 semi-quantitative scoring scale (details in Supplementary Methods). Mean scores were calculated for each individual kidney, and final results were expressed as group means ± SD.
Statistical Analysis: Results were expressed as means ± SD, and differences between conditions were tested statistically by ANOVA and post-hoc tests where indicated using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Significance was assigned at p < 0.05. Unsupervised hierarchical clustering and the generation of the corresponding heat map were performed using the Morpheus online visualization and analysis suite (Broad Institute, Cambridge, MA, https://software.broadinstitute.org/morpheus). All data, except gamma glutamyl-transpeptidase (Gamma GT) and albumin-globulin ratio (Albumin:Globulin), were subjected to log2 transformation in Excel (version 2013, Microsoft, Redmond, WA), prior to clustering. The hierarchical relationship of data patterns for all animals was establish by average linkage, Euclidean distance method.
{kind=link}