Full-length transcriptome for W. pigra
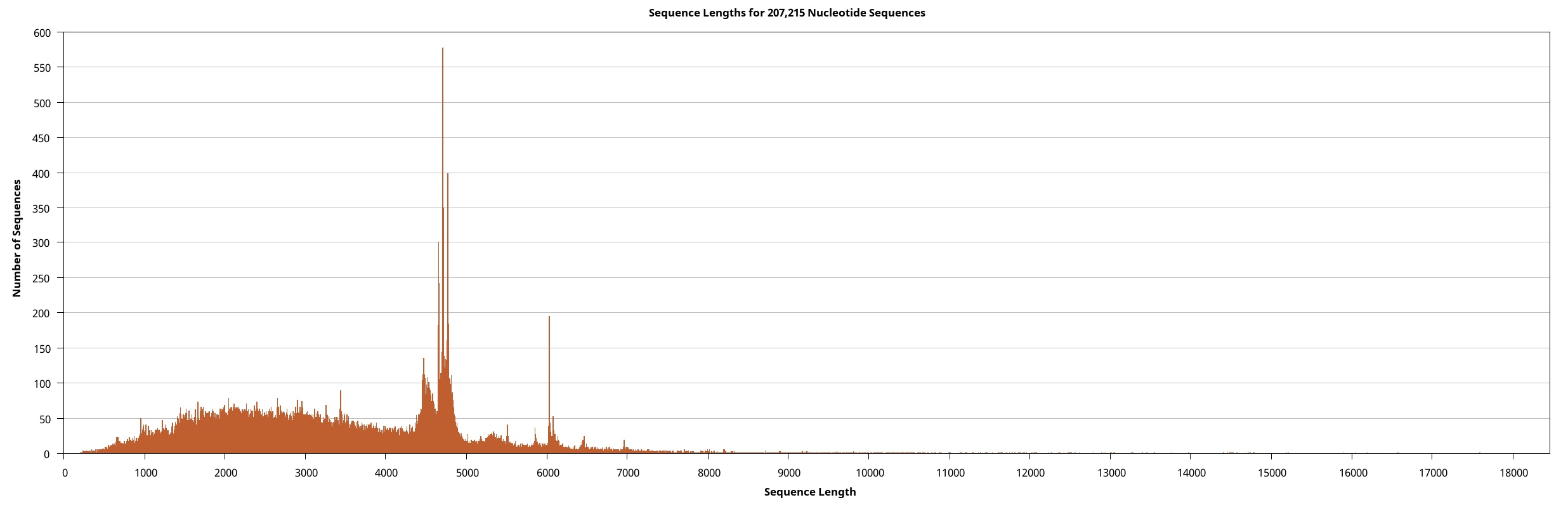
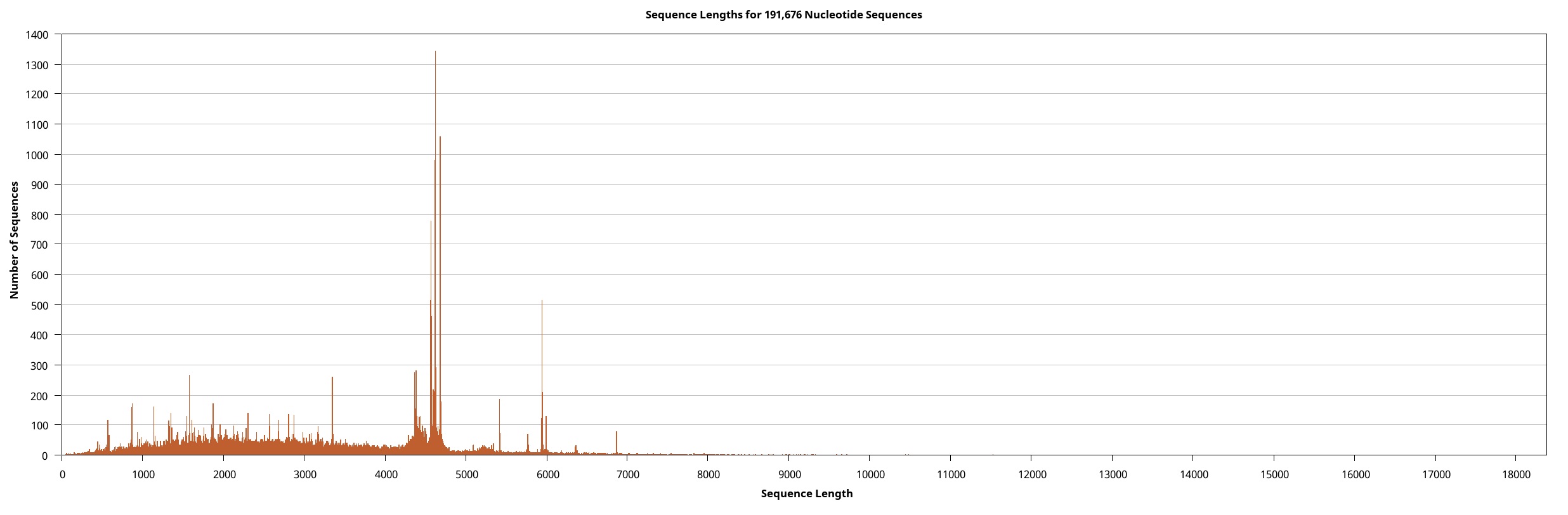
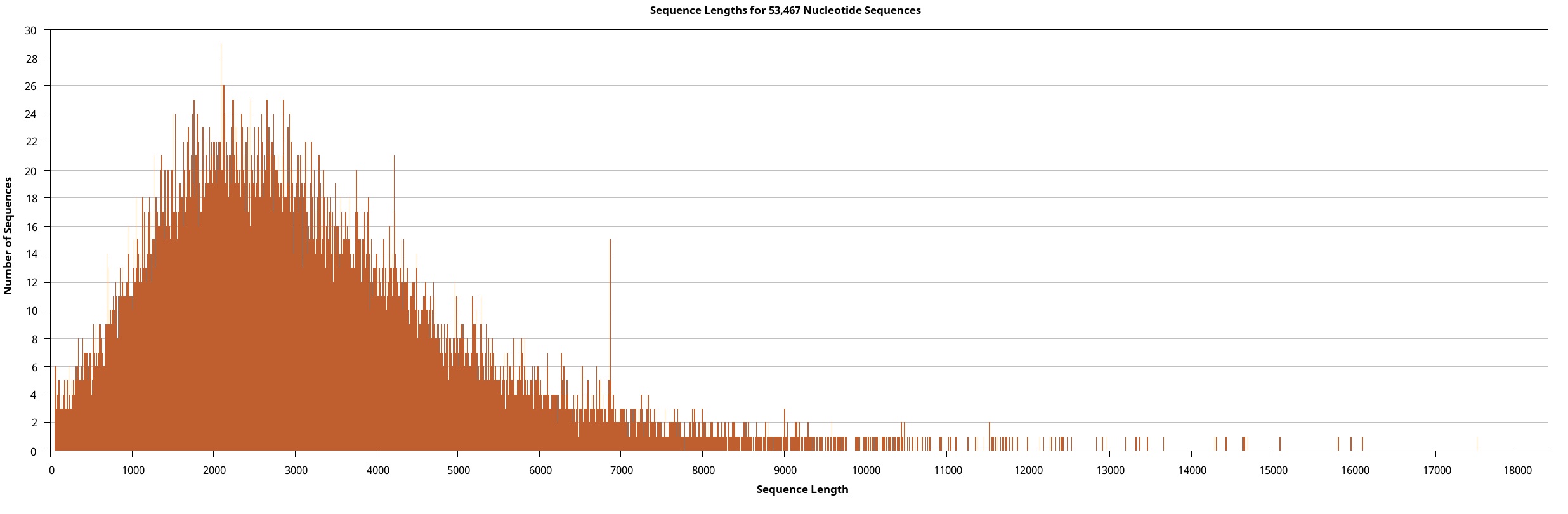
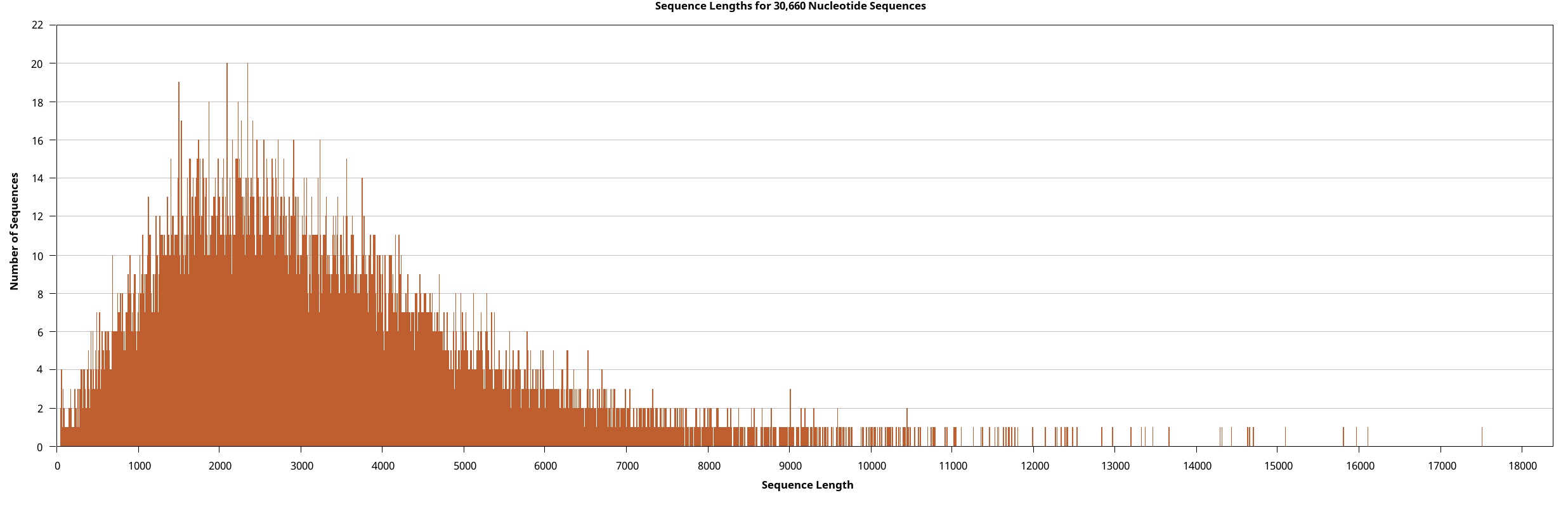
We generated the full-length transcriptome dataset of W. pigra using a pooled sample via the Pacbio platform. Overall, 207,215 circular consensus sequences (CCS) of 705.78 Mb, were retrieved. These CCSs were clustered and assembled into 191,676 (92.50%) non-redundant full-length non-chimeric reads (FLNC). All FLNCs (multiple copies of the same transcript) were then clustered. For each obtained cluster, a consensus isoform was generated, a total of 53,467 high-quality (HQ) isoforms with accuracy greater than 99% were obtained. Eventually, we precisely removed redundant sequences using the CD-HIT software and obtained 30,660 transcripts (a total length of 93.19 Mb). Table 1 outlines the statistical summary for lengths and sizes of CCS, FLNC, and HQ isoforms and transcripts. The length distributions of these sequences depict their consistency and quality (Supplement Fig. 1 ~ 4). In particular, the connectivity of transcripts was much higher than that of previously assembled transcriptomes (all are averagely below 1000 bp long) and is comparable to the high-quality genome of another medical leech, Hirudinaria manillensis (average length of 2963.1 bp). The transcripts served as the final molecular sequence pool for screening gene components and genetic motifs. Based on BUSCO (Bench-marking Universal Single-Copy Orthologues) completeness analysis, 74.8% of BUSCOs were complete, among which 25.8% were duplicates (Fig. 1A).
Table 1
Sequence statistics for full-length transcriptome of W.pigra.
|
File type
|
Nucleotides (bp)
|
Sequences
|
GC (%)
|
Min Sequence Length (bp)
|
Max Sequence Length (bp)
|
Mean length (bp)
|
Total Size
(Kb)
|
|
CCS
|
705,787,087
|
207,215
|
44.30
|
201
|
17590
|
3406.1
|
175,433
|
|
FLNC
|
619,526,806
|
191,676
|
46.01
|
50
|
17506
|
3232.2
|
154,138
|
|
HQ isoforms
|
159,075,749
|
53,467
|
43.30
|
50
|
17506
|
2975.2
|
39,733
|
|
Transcripts
|
93,199,769
|
30,660
|
42.40
|
52
|
17506
|
3039.8
|
23,348
|
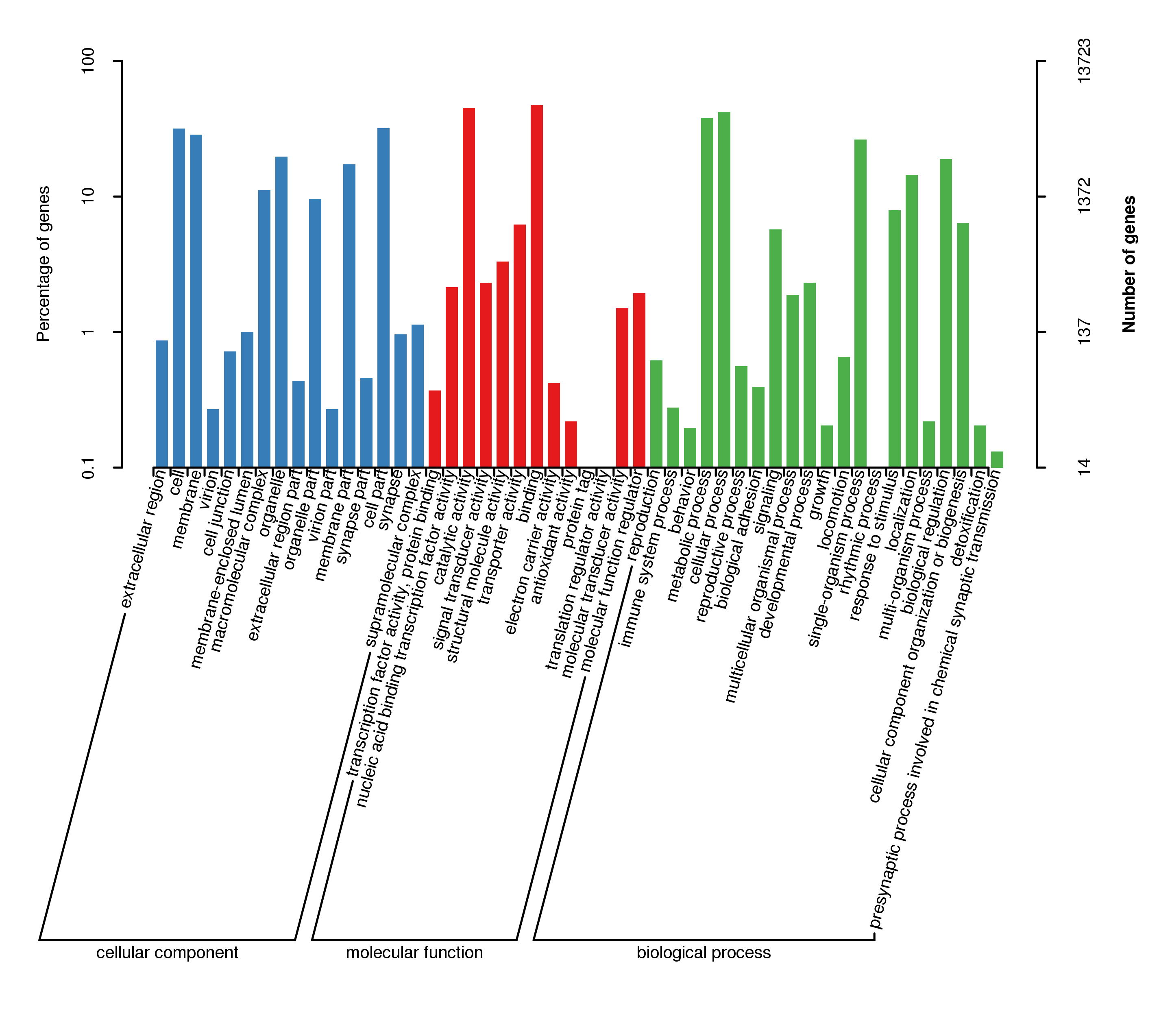
All HQ transcripts were annotated in public databases, including NT, NR, Pfam GO, COG/KOG, KEGG, Swiss-Prot, and eggnog (Table S1). We annotated 28,144 transcripts accounting for 91.79% of the total number. More transcripts were annotated in NR (27,737), followed by eggnog (25,433), and Pfam (24,538). The homologous species distribution of W. pigra annotated in the NR database demonstrated that its gene sequences exhibited high homology with several other Annelids, including the freshwater leech Helodbella robusta (16171, 58.38%), and the sea worm Capitella teleta (2401, 8.67%) (Fig. 1B). GO classifications assigned 13,723 transcripts into 50 specific GO terms. The categories with the largest number of transcripts in each classification were as follows: Molecular function (binding, 6,523), biological process (cellular process, 5,788), and cellular component (cell part, 4,384) (Supplement Fig. 5).
We identified 24,719 CDSs from all transcripts, with a total length of 36.47 Mb. Through a combination of four different prediction methods, the Coding-Non-Coding Index (CNCI) (Sun et al., 2013), the Coding Potential Calculator (CPC) (Kong et al., 2007), the Pfam-scan (Pfam) (Finn et al., 2016) and the Coding Potential Assessment Tool (CPAT) (Li et al., 2014), 1,314 LncRNAs were revealed (Fig. 1C). We successfully located the corresponding target CDSs for nearly all LncRNAs (1,308 out of 1,314). The total number of targets was 12,775. In total, 2,574 alternative splicing (AS) events were predicted, and 338 of these transcripts were subjected to several origins more than once. Also, 932 transcript factors (TFs) were predicted. The zf-C2H2 was the most abundant TF. In addition, 33,258 SSRs were found, which comprised 5,592 mononucleotides, 1,485 dinucleotides, 24,737 trinucleotides, 1,162 tetranucleotides, 100 pentanucleotides, and 182 hexanucleotides. The number of SSR-containing sequences was 11,478. All the data retrieved in this work, including the sequences of transcripts, along with the annotation information, genetic motifs (LncRNAs, SSR, TFs), predicted alternative splicing, and their target CDSs, served as solid genetic database for W.pigra, and were deposited in Zenodo using the link: https://doi.org/10.5281/zenodo.4707300.
Identification of anticoagulant genes
We searched for all known types of anticoagulant genes that have been reported in leeches from the Swiss-prot database according to the annotations of transcripts. A total of 426 transcripts met such criteria (Supplementary File S1). These transcripts were further assigned into 8 Pfam protein families, including the antistasin family (121 genes, PF02822.13), Destabilase (14 genes, PF05497.11), Kazal-type serpin (62 genes, PF07648.14), Lectin C (72 genes, PF00059.20), Fibrinogen beta and gamma chains C-terminal globular domain (124 genes, PF00147.17), Eglin C (12 genes, PF00280.17), Cystatin family (17 genes, PF00031.20) and Neurohemerythrin (5 genes, PF01814.22). Among them, the transcript sequences for 16 genes were novel, yielded by alternative-splicing events, including a member of the Antistasin family (JXZ_transcript_6437) exhibiting a direct anticoagulant effect. Its nucleotide sequence had a gap when compared to the origin sequence (JXZ_transcript_50726) but encoded the same protein (Supplement Figure S6).
Meanwhile, we found 25 transcripts with no blast hits in the NR and swiss-prot databases (Supplement Table S2). They were suggested to produce newly identified anticoagulant proteins, which complement the existing anticoagulant reservoir of leeches and have proven that although it has been studied for years, W. pigra still has the potential on digging functional components. The new anticoagulant proteins include 6 antistasins, 3 Kazal-type serpins, 13 Lectin C, and 3 Cystatins. The conserved domains for their translated proteins were illustrated using seqlogo maps to depict the composition and variation pattern of their sequences (Fig. 2). The seqlogo maps demonstrated that the proteins with Lectin C domains harbor the most complex variation in sequences, whereas members of the Antistasin family all comprise typical Cysteine-rich structures.
LncRNA regulation network for anticoagulant genes
LncRNAs are regulation factors for protein-coding sequences. In previous sections, we have predicted the target sequences for all 1,314 LncRNAs and revealed 12,775 targets. Their relationships will guide future genetic studies such as inhibition or activation of focused gene expressions through manipulation of specific LncRNAs. Based on the linkages between the LncRNAs and target transcripts, we have generated a linkage map for all 426 anticoagulant transcripts (Supplement Figure S7). Notably, we found a specific node linked to a single type of transcript, encoding the Guamerin protein. This node represents a regulation mechanism with highly abundant factors. We have illustrated its regulation network in Fig. 3. The elements of 3 LncRNAs (JXZ_transcripts_27429, JXZ_transcripts_39693, and JXZ_transcripts_15344) are shown with the typical many-to-many correspondence trend with the coding transcripts, which indicate their importance in this regulation mechanism.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}