The expression of the obestatin/GPR39 system in the peripheral nerve system. In adult rat sciatic nerve, GPR39 localized primarily to SCs of myelinated fibers, while obestatin showed a diffuse expression pattern (Fig. 1a). Strong GPR39 immunoreactivity was associated with the plasma membrane and ectoplasm of larger-sized DRG neurons, whereas more diffuse cytoplasmic immunostaining, with occasional granular staining of the plasma membrane, appeared in the smaller-sized DRG neurons (Fig. 1b). Diffuse cytoplasmic immunostaining was observed for obestatin in DRG neurons (Fig. 1b). In rat anterior horn of spinal cord, GPR39 immunostaining was observed in the motor neurons and glia, whereas obestatin immunoreactivity was only observed in motor neurons (Fig. 1c). The expression pattern of obestatin/GPR39 in PNS and, in particular in SCs, prompted us to examine its expression pattern in response to nerve injury. We initially performed a spatial analysis of the obestatin/GPR39 system on a recovered rat sciatic nerve after a complete transection (Fig. 1d). We found that, by 12d after the cut, proximal and distal compartments showed GPR39 positivity, which was strongly associated to longitudinal cell columns, the bands of Büngner, in the distal part (Fig. 1d). Of note, obestatin expression varied significantly between distal and proximal areas revealing an increase in the distal compared to proximal stump (Fig. 1d). Using cell-type-specific markers to identify obestatin-positive cells in the distal nerve stump by immunofluorescence, we identified SCs, Sox10 positives cells (Sox10+), expressing high levels of obestatin (Fig. 2). In contrast, macrophages (F4/80), endothelial cells (CD31) and regenerating axons (neurofilament, NF) in the distal nerve stump did not express levels of obestatin (Fig. 2), ruling out their implications as sources for the expression of this peptide in the distal nerve stump. Thus, nerve injury was associated with increased obestatin levels in the distal SCs consistent with a role as chemoattractant to guide cells and their accompanying axons out of the nerve stumps and across the bridge during peripheral nerve regeneration. We also confirmed obestatin expression on cultured IFRS1 cells (data not shown).
Obestatin signaling drives peripheral nerve regeneration. To assess the obestatin-related regenerative response in the peripheral nerve system we employed a standard compression model of sciatic nerve injury (axonotmesis), an injury model that allows to study the interaction among regenerative axon with both the SCs and basal laminae [40]. Obestatin (500 nM/Kg body weight per 48 h for 12d) or vehicle [0.9% NaCL (w/v), corresponding volume] was administered into crush-injured sciatic nerve by using a catheter port (Fig. 3a). Motor function was assessed with standard SFI analysis (see details in the Footprint Test from the Method section), using measurements of total footprint length (PL), toe spread (TS), and intermediate toe spread (ITS). Walking footprint patterns and quantification of SFI revealed functional recovery of obestatin-treated rats as compared to control rats at 3, 6 and 12d post-injury (Fig. 3b, right and left panels, respectively). At 21d post-injury, there was a significant increase in the expression of the neuronal outgrowth marker, growth-associated protein-43 (GAP43), in the crush and distal sites of obestatin-treated rats compared to control (Fig. 3c). Within 12d of injury, longitudinal sections of regenerating nerves exhibited a significant increase in the NF-stained positive axons (NF+) in the nerve segment distal to the crush site of the obestatin-treated nerves compared to control (Fig. 3d). There was also an increase for the SC marker, myelin basic protein (MBP) in the distal nerve segment (Fig. 3e). In this segment, the number of MBP+ longitudinal tubular structures were augmented noticeably in obestatin-treated rats at 12d post-injury, showing a decrease of ellipsoid bodies also termed as degeneration chambers (Fig. 3e). Consistently, immunoblot analysis demonstrated the upregulation of GAP43 and makers of myelinating SCs such as MBP, myelin-associated glycoprotein (MAG), myelin protein zero (MPZ), POU protein Oct6 (OCT6) and early growth response 2 (EGR2) in obestatin-treated rats relative to control animals (Fig. 3f). Together, these data imply that obestatin signaling favors the ability of dedifferentiated SCs to switch back to a myelinating phenotype on contact with regenerated axons.
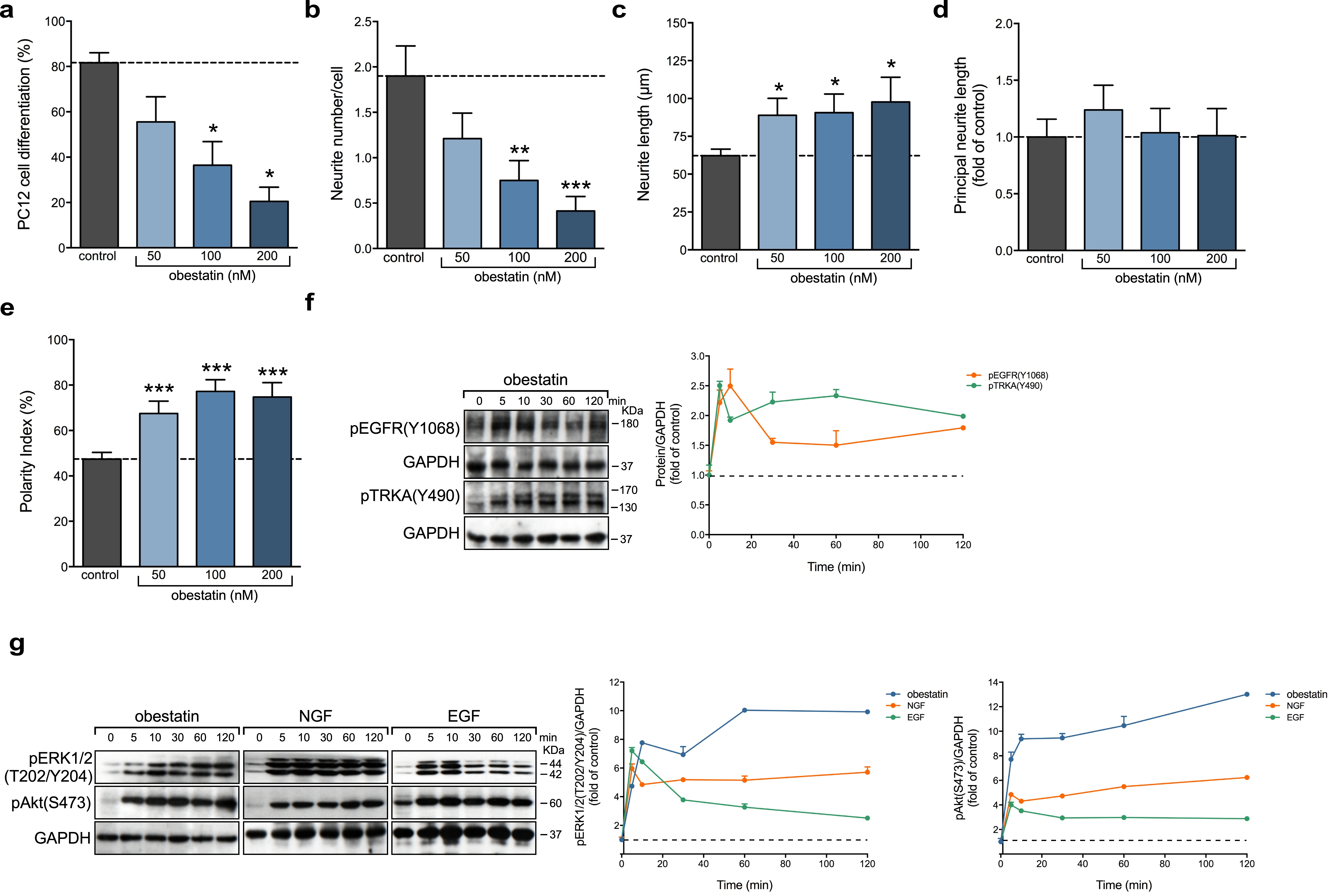
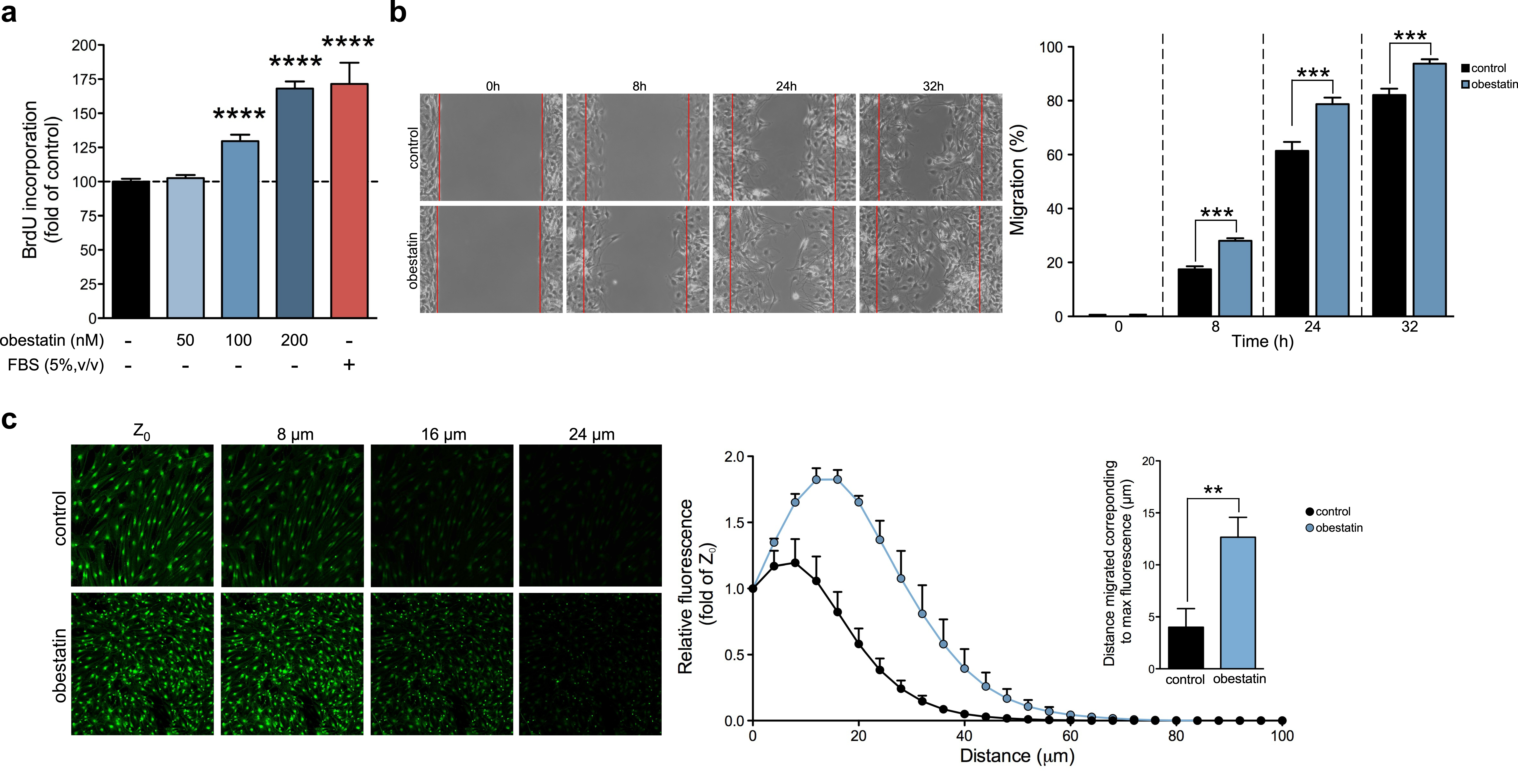
Because myelination and axonal integrity play a critical role in the functional recovery after PNS injury, the obestatin effects in these parameters was evaluated taking the advantage by using the immortalized rat SC, IFRS1, and the model of neuronal differentiation, PC12, cells. As shown in Figure S1a, obestatin led to a significant dose-dependent upregulation of IRFS1 proliferation rates when compared to control. Added to mitogenic effect, a wound-healing assay comparing the migration of IRFS1 cells at different time points revealed that obestatin acted as a mobility signal for SCs (Figure S1b). Furthermore, an inverted Boyden chamber migration assay supported the role of obestatin (200 nM) as chemotactic signal for SCs (Figure S1c). IFRS1 cells were able to migrate through the membrane, mimicking basement membrane invasion and invade into the Matrigel→ as an extracellular matrix when obestatin (200 nM) was applied on top of the MatrigelⓇ as a chemoattractant (Figure S1c). With respect to PC12 cells, the role of obestatin signaling was analyzed in differentiated PC12 cells (dPC12 cells: 7d DM-primed PC12 cells). Obestatin-treated dPC12 cells showed a reduction in the number of neurites in accordance with the decrease in the percentage of differentiated cells (Figure S2a and S2b, respectively). Of note, obestatin increased mean neurite length (Figure S2c) and polarity index (Figure S2e), with no significant effect on principal neurite length (Figure S2d) by promoting the conversion from the multipolar morphology to the bipolar one. These observations were consistent with the early signaling pathways activated by obestatin/GPR39 system in PC12 cells. It is known that epidermal growth factor (EGF) and NGF show opposing actions to proliferate or differentiate which are dictated by the duration of extracellular signal regulated kinase 1/2 (ERK1/2) signaling in PC12 cells (41). When PC12 cells where stimulated with obestatin (200 nM) led to transient phosphorylation of the EGF receptor (EGFR) at Y1068 and sustained phosphorylation response of NGF receptor (TRKA) at Y490 (Figure S2f). The interplay among G protein-coupled GPR39, EGFR and TRKA activated by obestatin signaling network triggered sustained ERK1/2 phosphorylation at T202/Y204 [pERK1/2(T202/Y204)] (Figure S2g). Similarly, obestatin (200 nM) evoked sustained Akt phosphorylation at S473 [pAkt(S473)] (Figure S2g). This temporal specification rewires the system toward cell differentiation in response to obestatin signaling. This observation further supports the importance of activation duration to yield the correct cell fate decision [41–47].
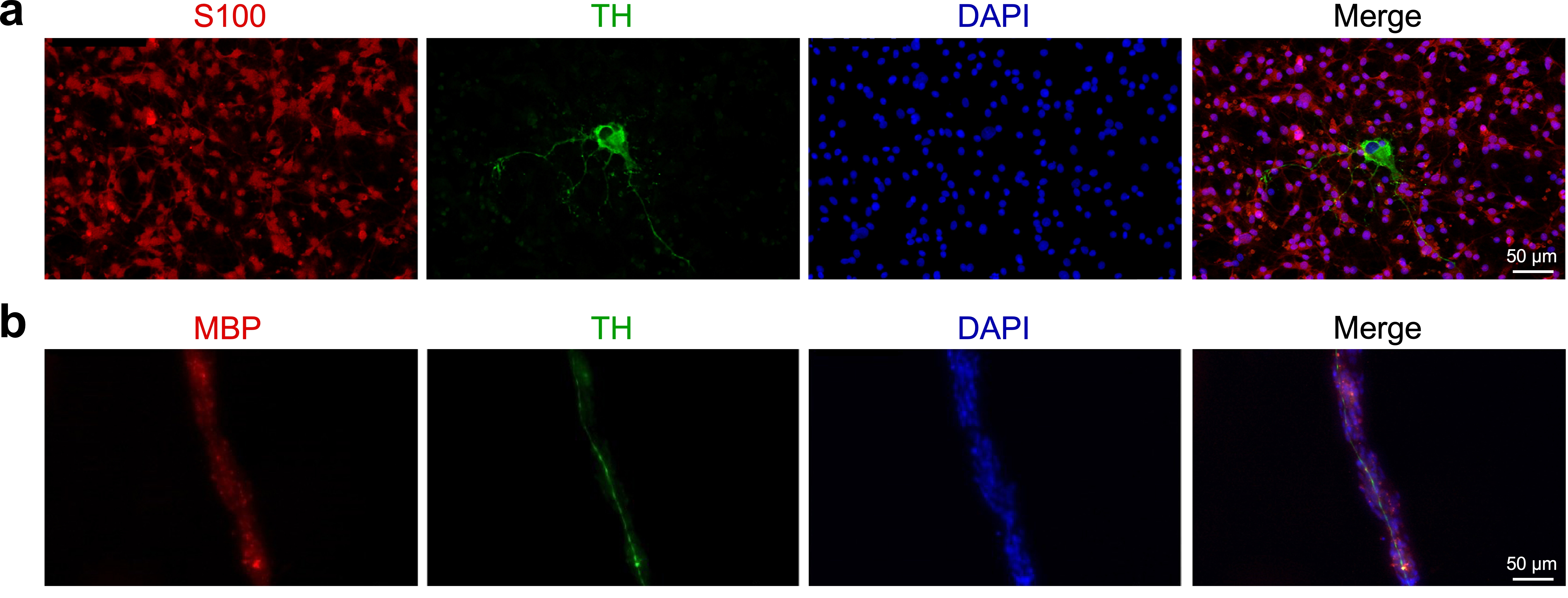
The role of the obestatin/GPR39 system in the interactions between SCs and neurons was stablished using a co-culture system that combine the SC cell line IFRS1 and the NGF-primed PC12 (33). In this PC12-IFRS1 model, PC12 cells acquired a neuronal phenotype, demonstrated by morphological activation with neurite elongation in close contact with SCs (Figure S3a). Importantly, MBP staining was mainly detected in areas corresponding to IRFS1 cells closely attached to the neurites emerging from PC12 cells, demonstrating a stable and effective SC-axon interaction (Figure S3b). In this model, cell proliferation assay showed a significant increase in obestatin-treated cells under myelinating conditions (200 nM; Fig. 4a). To discriminate between IRFS1 and PC12 cell proliferation, both cell types were cultured for 7d in myelinating conditions and the proliferation was analyzed separately. As expected, obestatin (200 nM) led to IRFS1 cell proliferation (Fig. 4b), with no evidence in PC12 cells (Fig. 4c). Additionally, we observed a marked increase in the number of IRFS1 cell clusters in the obestatin-treated co-culture compared to control (200 nM; Fig. 4d). Analysis of cluster area revealed an increase in obestatin-treated co-culture (Fig. 4e). Likewise, transect measurements across clusters revealed an increase in cell density in obestatin-treated co-cultures compared to control (Fig. 4f). Finally, analysis of several key myelin proteins was supportive of a global increase in myelinization levels upon obestatin treatment. In particular, obestatin increased the expression of MPZ, MBP and MAG in IFRS1 cells within 21d after coculture (Fig. 4g). These changes were directly correlated with the increase in the lipid biosynthesis and the expression of sterol regulatory element-binding protein (SREBP) cleavage activating protein (SCAP) (Fig. 3h and 3i, respectively) at 21d post-co-culture. High levels of MBP were evident in obestatin-treated co-culture with colocalization to MBP+-SCs and tyrosine hydrolase (TH)-stained axons (Fig. 4j). This result was further analyzed by Manders m2 coefficient, where an increase in overlap of PC12 cells by SCs was evident in obestatin-treated group compared with control (Fig. 4j). Accordingly, the transects taken perpendicularly along PC12 neurites demonstrated an increment of the MBP+-SC density in the obestatin-treated group compared to control (Fig. 4k). Taken together, these findings support the role that obestatin signaling exerts in the myelination and axonal integrity.
Obestatin-mediated regeneration of sciatic nerve reverses skeletal muscle atrophy. Since decline in neuromuscular innervation is known as an endogenous cause of muscle atrophy, we first measured the TA, extensor digitorum longus (EDL), gastrocnemius (GM) and soleus muscle weights to assess muscle atrophy. 21d after injury, we observed loss of muscle mass in the control group (sciatic nerve injury control) that was counteracted by obestatin treatment in all tested muscles (Fig. 5a). Using HE stained cross sections of the TA muscles from each group fiber, cross-sectional areas (CSA) were measured and compared among groups (Fig. 5b). Remarkable, CSA analysis showed that the treated-to-control ratio of all fibbers was 43% larger in obestatin-treated group (3665 ± 49 µm2) than in control (2564 ± 35 µm2) at 21d post-injury (Fig. 5b). The myofiber area distribution showed that high percentage of the individual fibers in the obestatin-treated group had fiber areas between 2500–3250 µm2, whereas most of the fibers in the control group had areas between 1500–2250 µm2 (Fig. 5b). In terms of protein degradation, obestatin treatment led to a significant decrease in the expression of the ubiquitin E3-ligases MAFbx and MuRF1 (Fig. 5c). The two major signaling pathways regulating skeletal muscle atrophy program are the Forkhead box O (FoxO) transcription factors and histone deacetylase proteins (HDAC) in several pathophysiological conditions, including neurogenic atrophy, muscle disuse, and cancer cachexia [48, 49]. In fact, the down-regulation of MAFbx and MuRF1 expression was concomitant with increased phosphorylation of FoxO3a at T32 and FoxO1 at T24 but did not change basal FoxO4 phosphorylation at T28 in the obestatin-treated group (Fig. 5d). However, the upregulation of HDAC4, which represses Dach2, a negative regulator of myogenin, resulted in myogenin expression in both obestatin and control groups (Fig. 5d) ruling out its implication in this proteolytic pathway. It is known the role that the mTORC1 pathway plays in stimulating protein synthesis in skeletal muscle [49]. After 21d of nerve injury, levels of the phosphorylated form of mTOR at S2448 increased in TA muscle in both control and obestatin-treated groups (Fig. 5e). We them focused our attention on the two best-characterized mTORC1 targets, the ribosomal protein S6 [downstream target of the serine/threonine kinase p70S6K1 (S6K1)], and the eIF4E-binding protein 1 (4E-BP1) [50, 51]. Levels of phosphorylated form of S6 at S240/244 were increased in control group in TA muscle, but this effect was significantly lower in obestatin-treated animals (Fig. 5e). Interestingly, an analysis of the phospho-forms of 4E-BP1 showed a strong increase in the hyperphosphorylated form of 4E-BP1, designated as g form, at T37/46 residues in obestatin-treated group, but not in control group (Fig. 5e). The interplay between the ubiquitin-proteasome and autophagy-lysosome systems determines the regulation proteostasis as well as its extent in the context of different catabolic or anabolic conditions [52]. In this particular case, autophagy induction was reduced in TA muscles in obestatin-treated animals, as shown by limited increase in the lipid modified form of LC3, referred as LC3II, and increased levels of p62 and cathepsin-L (mature form), in relation to control group (Fig. 5f). Taken together, these data support a model whereby the interplay between mTOR and FoxO regulates the ubiquitin-proteasome and the autophagy-lysosome systems, and the signaling associated with protein translation in response to the obestatin/GPR39 system in the target muscle of the regenerating nerve.
Obestatin signaling delays axonal degeneration and neuromuscular synaptic loss upon nerve injury. To test if the function of obestatin in preventing muscle wasting was related to the inhibition of axonal degeneration, we analyzed proteins involved in mitogen-activated protein kinase (MAPK)/glycogen synthase kinase 3β (GSK3β) signaling, apoptosis, and cytoskeleton formation in the sciatic nerve (Fig. 6a). Compared to control sciatic nerves, pERK1/2(T202/Y204), a pathway classically associated with neurite outgrowth [53, 54], was significantly increased in obestatin-treated sciatic nerves at 12d post-injury (Fig. 6a). The activation of ERK1/2 was concurrent to the inactivation of GSK3β, estimated as GSK3β phosphorylation at S21/9 [pGSK3β(S21/9); Fig. 6a], a pathway required for transforming neurons into a regenerative state upon injury [55]. Remarkably, obestatin-treated sciatic nerves exhibited significant increase of c-Jun-N-terminal kinase (JNK) phosphorylation at T183/Y185 [pJNK(T183/Y185)], a signaling node involved in axonal growth and regeneration (Fig. 6a) [56]. Unexpectedly, the proapoptotic BH3-only protein Bim and the apoptosis regulator Bax were increased in response to obestatin signaling, suggestive of apoptosis (Fig. 6a). However, the cleaved and active caspase 3 expression, major effector in neurite degeneration, was clearly downregulated in obestatin-treated group, especially when compared with control group (Fig. 6a). Despite the activation of these apoptosome pathway components, the survival promoting kinases associated to obestatin signaling are effective enough for regulating caspase 3 expression and thus inhibiting neuronal apoptosis. Finally, obestatin-treated group showed significantly increase of axonal proteins such as NF medium (NF-M) and NF light (NF-L), with significantly upregulation of a-tubulin and ßIII-tubulin (Fig. 6a). NFs not only provide structural support for neurons, but also interacts with many proteins and organelles, including tubulin, to establish a regionally specialized network that serves as a docking platform to organize other organelles and proteins [57]. These results imply that obestatin may protect neuromuscular synapses through mechanisms involving the inhibition of axonal degeneration.
Calpastatin, an endogenous inhibitor of calcium-dependent cysteine protease calpain, is involved in protein degradation, neuromuscular function regulation [38], and axon survival [58]. A noteworthy increase of the calpastatin level was observed in obestatin-treated sciatic nerves 12d post-injury (Fig. 6b). Interestingly, the change of calpastatin level correlated with upregulation of the calpain-2 level in the obestatin-treated group, while calpain-1 remained unchanged (Fig. 6b). In contrast, the calpain-1 level was increased in the control group, whereas calpastatin and calpain-2 remained unchanged (Fig. 6b). In both cases, the calpastatin level and its balance with calpain activity were key determinants of how calpains are regulated. Additionally, obestatin-treated sciatic nerves exhibited significant increase in the mitochondrial outer membrane protein Mfn2 levels, a protein involved in the axonal transport of calpastatin to protect NMJs [38]. Mfn2 is enriched at the junction between the endoplasmic reticulum (ER) and mitochondria, which is known as the mitochondria-associated ER membranes (MAMs) [59]. Consistent with Mfn2 findings, an increase in the ER marker calnexin level was noted in obestatin-treated sciatic nerves (Fig. 6b). Thus, Mfn2 upregulation delayed onset and progression in this model of sciatic nerve injury by raising calpastatin levels, essential for axonal survival. Indeed, the change of calpastatin levels correlated with the cytoskeletal protein levels, specifically NF-M, NF-L, ßIII-tubulin and a-tubulin. Additionally, the nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1) levels showed to be regulated by obestatin signaling (Fig. 6b) supporting the view that Nmnat1 inhibits an upstream step leading to calpastatin depletion.
The role of Mfn2 and mitochondria in the axonal transport of calpastatin is sufficient to inhibit localized calpain activation, axon degradation, neuromuscular synaptic loss and muscle atrophy upon nerve injury [38]. Remarkably, the ratio of acetylcholine receptor (AChR)-rich postsynaptic sites on myofibers was increased in TA muscles after sciatic nerve injury under obestatin administration (Fig. 7a). Agrin, muscle-specific kinase (MuSK), and Wnt family member 3 (Wnt3) are key regulators of NMJs. Agrin and Wnt3 are secreted by motor neurons, whereas MuSK is mainly expressed in skeletal muscles [60]. In skeletal muscles, obestatin-treated rats exhibited significant increase in the expression levels of Agrin and Musk proteins but not Wnt3 protein at 12d post-injury (Fig. 7b). In sciatic nerve, obestatin-treated rats showed significant increased levels of Agrin and Wnt3 proteins related to control (Figs. 7b). Remarkably, in rats with sciatic nerve injury under obestatin administration, NMJ innervation, estimated by Pearson and Manders correlation coefficients between synaptic vesicle glycoprotein 2A (SV2) and AchR (a-bungarotoxin for motor endplates), was sustained at a level comparable with this of rats with no sciatic nerve injury (Fig. 7c). These data provide evidence supporting the role of obestatin signaling in preserving neuromuscular synapse loss upon nerve injury through the regulation of Mfn2-mediated calpastatin transport and, thus, the calpain-calpastatin proteolytic system.
{kind=link}
{kind=link}
{kind=link}