3.1 Geometries and adsorption of species in FA decomposition
For the possible species in FA decomposition, the obtained most stable configurations are presented in Fig. 2. The corresponding adsorption energies and selected bond distances are listed in Table 1.
HCOOH. The reaction of FA decomposition starts with the adsorption of gaseous HCOOH. After it is adsorbed on the Pd surface, two different isomers have been obtained, i.e., trans-HCOOH* and cis-HCOOH* (Fig. 2). For trans-HCOOH*, the H atom in hydroxyl group (OH) points toward the bridge site on the Pd surface and trans-HCOOH* bonds with Pd atom at the top site by another O atom with the Pd-O bond distance of 2.33 Å, in agreement with the previous study [17]. For cis-HCOOH*, it also bonds with Pd atom at the tope site with the Pd-O bond distance of 2.58 Å, but the H atom in OH pointing away from the catalyst surface. The adsorption energy for trans-HCOOH* is -0.71 eV (Table 1), stronger than -0.62 eV by PW91 [14] and -0.37 eV by PBE [17]. This could be caused by the different theoretical methods or the vdW interaction considered in this work, or both. For cis-HCOOH*, the adsorption energy is weak (-0.34 eV) compared with the trans-HCOOH*. This value (-0.34 eV) is stronger than -0.11 eV by PBE [17], but weaker than -0.41 eV by PW91 [14]. In addition, the parallel adsorption structure of HCOOH* with Pd surface in the previous study [15, 36] has also been taken into account, in which HCOOH* bonds with the surface through C, O atoms. But unfortunately, we failed to locate this structure.
Table 1 Adsorption sites, bond distances (Å) and adsorption energies (eV) for the most stable adsorbed species in formic acid decomposition on the Pd(111) surface. Comparison is made with the previous studies.
| |
RPBE
|
PBE a
|
PW91 b
|
|
site
|
∆Eads
|
dPd-C/H
|
dPd-O
|
site
|
∆Eads
|
site
|
∆Eads
|
|
trans-HCOOH
|
top
|
-0.71
|
|
2.33
|
top
|
-0.37
|
top
|
-0.62
|
|
cis-HCOOH
|
top
|
-0.34
|
|
2.58
|
top
|
-0.11
|
top
|
-0.41
|
|
bi-HCOO
|
bri
|
-2.58
|
|
2.17, 2.17
|
bri
|
-2.32
|
bri
|
-2.70
|
|
mono-HCOO
|
top
|
-1.92
|
1.97
|
2.10
|
top
|
-1.54
|
top
|
-2.04
|
|
trans-COOH
|
top
|
-2.55
|
2.00
|
|
bri
|
-2.16
|
top
|
-2.57
|
|
cis-COOH
|
top
|
-2.38
|
1.99
|
|
bri
|
-2.15
|
top
|
-2.41
|
|
CO2
|
bri
|
-0.19
|
|
3.22,3.22
|
bri
|
-0.02
|
bri
|
-0.18
|
|
CO
|
fcc
|
-2.00
|
2.02, 2.06, 2.08
|
|
fcc
|
-2.04
|
fcc
|
-2.05
|
|
H
|
fcc
|
-2.82
|
1.81
|
|
fcc
|
-2.87
|
fcc
|
-2.91
|
|
OH
|
bri
|
-2.44
|
|
2.15 ,2.17
|
bri
|
-2.57
|
bri
|
-2.62
|
|
H2O
|
top
|
-0.31
|
|
2.51
|
top
|
-0.25
|
top
|
-0.51
|
a Ref. [17] and b Ref. [14]
HCOO. HCOO* is the main intermediate in FA decomposition. It bonds with the Pd surface and forms two isomers, i.e., bi-dentate HCOO* (bi-HCOO*) and mono-dentate HCOO* (mono-HCOO*). For bi-HCOO*, both O atoms bonds with Pd surface with the same bond distance of 2.17 Å (Fig. 2 and Table 1). The adsorption energy is -2.58 eV, indicating a very strong adsorption. For mono-HCOO*, only the O atom at the carbonyl group (C=O) bonds with the Pd atom with Pd-O bond distance of 2.10 Å. Meanwhile, the H atom in C-H bond of mono-HCOO* points toward the atop Pd atom with a distance of 1.97 Å. The adsorption energy is -1.92 eV, much weaker than -2.58 eV for bi-HCOO*, consistent with the previous studies [14,17].
COOH. Similar to HCOO*, there are also two isomers for COOH*, i.e., trans-COOH* and cis-COOH*. For both configurations, only C atom bonds with Pd surface with the bond distances of 2.00 Å for trans-COOH* and 1.99 Å for cis-COOH*. For trans-COOH*, the H atom points away from the Pd surface with the adsorption energy of -2.55 eV. For cis-COOH*, the H atom points toward the Pd surface with the adsorption energy of -2.38 eV, weaker than trans-COOH*. From Table 1, we also noted that in this study and previous study by PW91 [14], both COOH* isomers adsorbed on the Pd top site, while they are adsorbed at the bridge site from the PBE study with nearly the same adsorption energies [17].
CO2. For adsorbed CO2, O-C-O bond angle is 179.3°, slightly deviated from the linear structure. The C=O bond distance is 1.18 Å, nearly the same as in the gaseous CO2 molecule, suggesting that CO2 is adsorbed physically on the surface. The adsorption energy is -0.19 eV, similar to -0.18 eV by PW91 [14], but stronger than -0.02 eV by PBE study [17].
CO. CO* is adsorbed on the fcc site with three slightly different Pd-C bond distances (Table 1). This is consistent with the experimental observation that CO* occupied hollow site by scanning tunneling microscopy [37]. The adsorption energy is -2.00 eV, similar to -2.05 eV by PW91 [14] and -2.04 eV by PBE [17].
H. H* atom can be stably adsorbed at the fcc site on Pd surface. This is in agreement with the experimental observation by low-energy electron diffraction that H* atom resides at fcc 3-fold hollow site on Pd(111) surface [38]. The calculated adsorption energy is -2.82 eV, also close to experimental value of -2.68 eV [39].
OH. For OH*, it prefers to bind at the bridge site with the adsorption energy of -2.44 eV. The two Pd-O bond distances are slightly different, i.e., 2.15 Å and 2.17 Å, respectively.
H2O. H2O interacts weakly with the Pd surface with the Pd-O distance of 2.51 Å. The adsorption energy is -0.31 eV, weaker than -0.51 eV by PW91 [14], but slightly stronger than -0.25 eV by PBE [17].
By comparing different methods, we noted that the adsorption energies from RPBE in this study lie between PBE and PW91 for cis-HCOOH*, bi-HCOO*, mono-HCOO*, trans-COOH*, cis-COOH* and H2O (Table 1). For trans-HCOOH* and CO2, the adsorption energies from RPBE are the strongest, while for CO*, H*, and OH*, RPBE gives the weakest adsorption energies.
3.2 FA decomposition mechanism
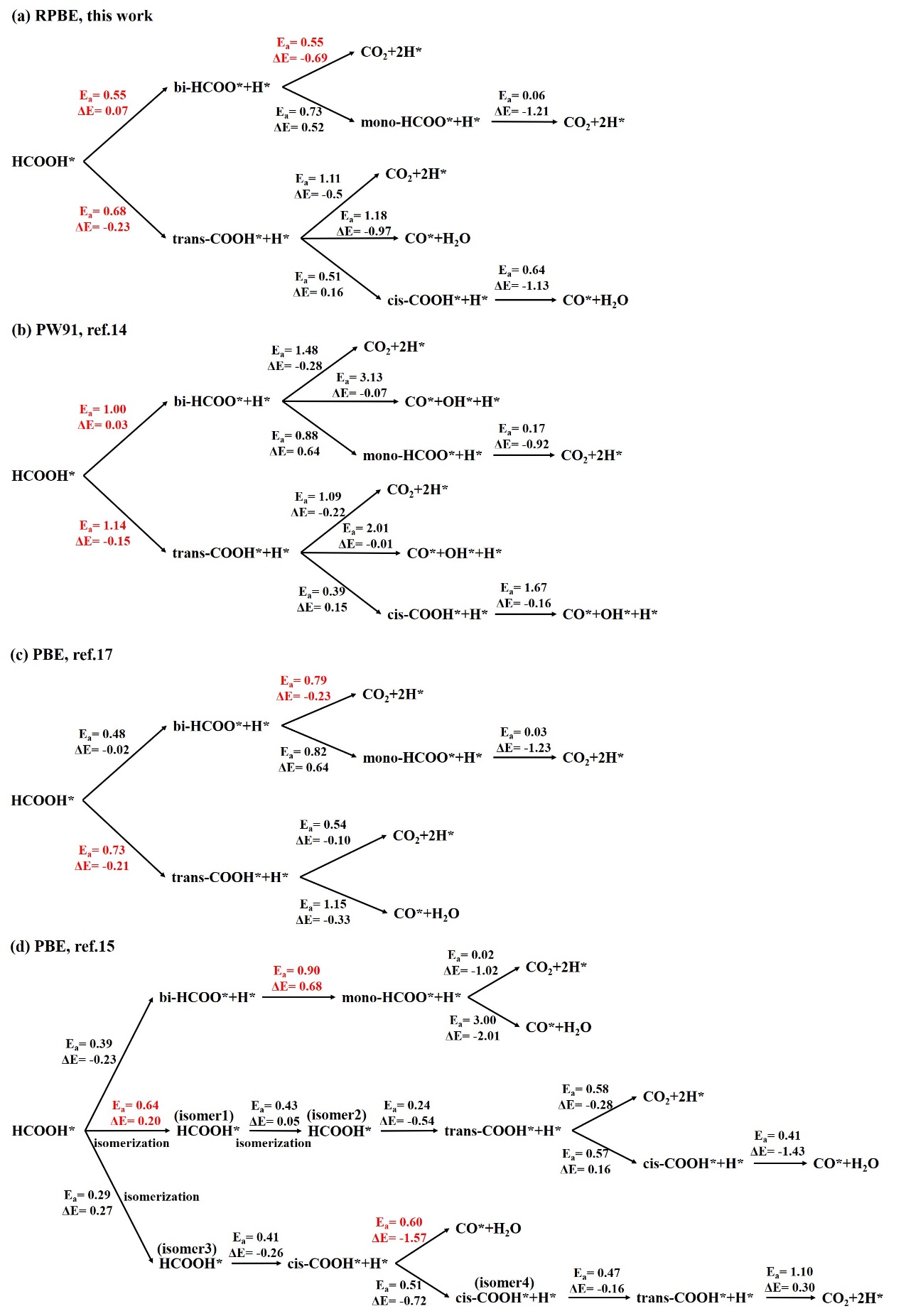
After obtaining the most stable adsorption structures, we can now procced to study the FA decomposition mechanism. The corresponding results are shown in Fig. 3 and Fig. 4. For comparison, our results are also presented in Scheme 1 together with previous theoretical studies.
FA decomposition via O-H bond cleavage (Fig. 3). In this mechanism, the first step is the FA dehydrogenation via O-H bond cleavage to form bi-HCOO*. In this step, the O-H bond in HCOOH* is elongated from the initial distance of 1.02 Å to 1.78 Å in transition state TS1. The energy barrier is 0.55 eV with slightly endothermic by 0.07 eV. We know that for isolated bi-HCOO* on the Pd surface, both Pd-O bond distances are the same (Table 1). However, for the complex state bi-HCOO* + H*, the existence of H atom makes the two Pd-O bonds having different distances, i.e., 2.16 and 2.23 Å, respectively. Subsequently, the longer Pd-O bond (2.23 Å) would be broken and the HCOO* species is rotated to form mono-HCOO*, in which the H atom in C-H group points toward Pd surface, which would be beneficial for C-H breaking. The reaction energy barrier in this step is 0.73 eV, which is also the rate-determining step. After the formation of mono-HCOO*, the formation of final product of CO2+2H* is very easy due to the negligible energy barrier of 0.06 eV. Thus, for the pathway trans-HCOOH* → bi-HCOO*+H* → mono-HCOO*+H* → CO2+2H*, the rate determining step is from bi-HCOO* to mono-HCOO*, similar to the previous study by PBE [15,17] (see also Scheme 1). For the other pathway, i.e., trans-HCOOH* → bi-HCOO*+H* → CO2+2H*, CO2 is produced directly from bi-HCOO*. In this pathway, the direct decomposition of bi-HCOO* is carried out through the cleavage of the longer Pd-O bond and the torsional deformation to form TS4, in which H atom in C-H is close to Pd surface. The energy barrier is 0.55 eV and the reaction is exothermic by -0.69 eV. This means that in the pathway trans-HCOOH* → bi-HCOO*+H* → CO2+2H*, both steps have the same energy barrier of 0.55 eV and are the rate-determining steps. This indicated that for the FA decomposition via O-H bond cleavage, pathway trans-HCOOH* → bi-HCOO*+H* → CO2+2H* is preferred. This is in agreement with the previous study by PBE [17].
FA decomposition via C-H bond cleavage (Fig. 4). The C-H bond cleavage in trans-HCOOH* would produce the intermediate trans-COOH*. The energy barrier is 0.68 eV. After trans-COOH* is formed, there are three possible pathways to produce the final products, i.e., trans-COOH*+H* → CO2+2H*, trans-COOH*+H* → CO*+H2O, and trans-COOH*+H* → cis-COOH*+H* → CO*+H2O (Fig. 4). For trans-COOH* directly converts to CO2+2H*, the energy barrier is 1.11 eV and exothermic by -0.50 eV. For trans-COOH* converts to CO*+H2O, the energy barrier is similar, i.e., 1.18 eV. This suggested that the direct conversion of trans-COOH* to the final product is difficult due to the high energy barriers. In contrast, the pathway trans-COOH*+H* → cis-COOH*+H* → CO*+H2O has relatively low energy barriers. The energy barrier is 0.51 eV from trans-COOH* to cis-COOH*, 0.64 eV from cis-COOH*+H* to CO*+H2O, which is more favorable. Thus, for FA decomposition via C-H bond cleavage, pathway trans-HCOOH* → trans-COOH*+H* → cis-COOH*+H* → CO*+H2O is favorable. The rate determining step is the formation of trans-COOH* from trans-HCOOH* with the energy barrier of 0.68 eV, in agreement with the previous study by PBE [17].
In a word, the above study showed that the FA decomposition via O-H bond cleavage is favored with the pathway of trans-HCOOH* → bi-HCOO*+H* → CO2+2H*. The energy barrier is 0.55 eV at the rate determining step, lower than 0.68 eV in the pathway of trans-HCOOH*→ trans-COOH*+H* → cis-COOH*+H* → CO*+H2O for CO production (Scheme 1). This conclusion is in agreement with the previous study by PBE [17], but different from the study by PW91 [14], in which the most favorable pathway is trans-HCOOH* → bi-HCOO*+H* → mono-HCOO*+H* → CO2+2H* (Scheme 1). Meanwhile, we also noted that in another study by PBE [15], the most favorable pathway is trans-HCOOH* → cis-COOH*+H* → CO*+H2O, while the most favorable pathway in this work, i.e., trans-HCOOH* → bi-HCOO*+H* → CO2+2H*, is not examined in Ref. 15. This demonstrated that computational methods would have great influence on both the geometries and electronic properties, and care should be taken in choosing the computational methods, for instance, calibrating the theoretical methods by comparing with the experimental study or more reliable theoretical methods. In addition, since the same method gives different conclusions, this remind us that we should be very careful in selecting the initial and final structures during the reaction.
Finally, from Scheme 1, we have also found something in common. All these theoretical studies showed that the formation of bi-HCOO* from HCOOH* is more favorable compared to the formation of trans-COOH* (HCOOH* → bi-HCOO* vs HCOOH* → trans-COOH*). Dehydrogenation pathway is favored along bi-HCOO* pathway, while dehydration pathway prefers to follow trans-COOH*. The formation of CO2+2H* from mono-HCOO (mono-HCOO* → CO2+2H*) is very easy due to the negligible energy barrier. This implies that mono-HCOO* can not exist stably on the catalyst surface because it will be dissociated immediately to the final product once it is formed. For the formation of CO*+H2O, the main intermediate is cis-COOH* and trans-COOH* is relatively easy to be transformed to cis-COOH*, while direct formation of CO*+H2O from trans-COOH* is difficult. This means that if more HCOO* species can be obtained experimentally and meanwhile reduce the formation of COOH*, the production yield of hydrogen would be high.
{kind=link}