Subjects
A total of 168 core families with probands diagnosed with ASD were recruited from the outpatient of Department of the Child and Adolescent Psychiatry, Shanghai Mental Health Center. All patients were diagnosed on the basis of the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V). All patients were Han Chinese and their age was ranged from 2 to 18 years.
Human H9 embryonic stem cells (hESCs) and intronic mutation (8-61757392-C-T) hESCs
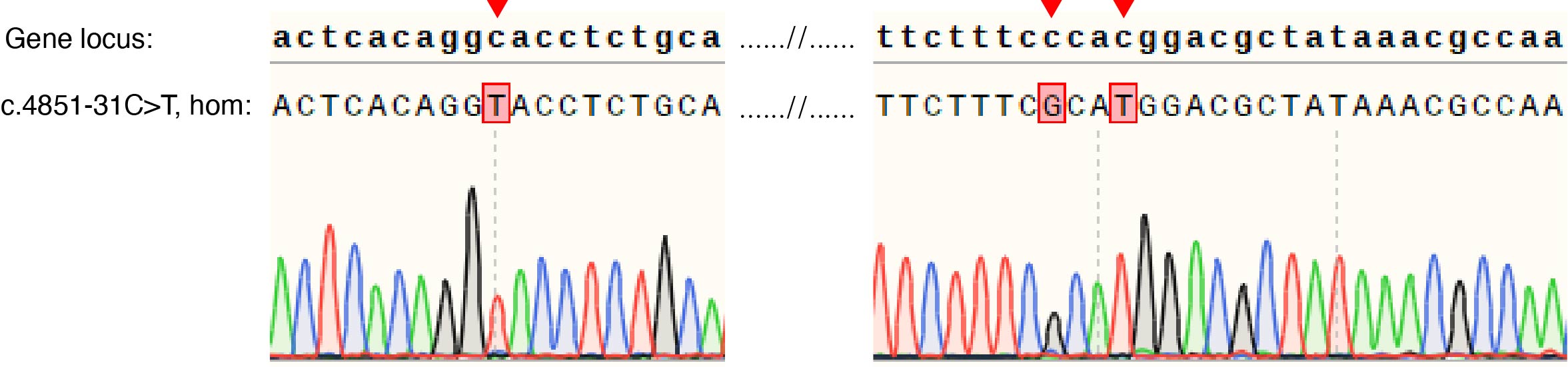
hESCs (H9, line WA09 (WiCell), passages 20-40) were cultured on a feeder layer of irradiated mouse embryonic fibroblasts (MEFs) in a humidified incubator with 5%CO2 at 37℃. The hESC medium containing DMEF/F12, 20% Knock Serum Replacement (Life Technologies), 0.1mM beta-mercaptoethanol (Sigma), 1% (v/v) Non-Eessential Amino Acids (NEAA, Life Technologies), 0.5% (v/v) GlutaMAX (Life Technologies). Medium was changed every day with 10ng/ml of bFGF (PeproTech). hESCs were passaged every 7 days. Intronic mutation (8-61757392-C-T) hESCs were transformation from H9 hESCs by introducing a intronic mutation and two synonymous mutations by homology-mediated end joining-based targeted integration using CRISPR/Cas9. The editor vector (addgene #48138) linked with sgRNA targeting sequence and the donor vetor homology with gene locus about 1.6kb exclude a intronic mutation and two synonymous mutations and flanked with sgRNA targeting sequence linked in pUC57 were liposome transfection by 1:1 ratio into H9 hESCs. Single GFP+ cells were isolated using flow cytometry and cultured. The intronic point mutation cells were identified by DNA sequencing.
HEK293 cells and HEK293 intronic mutation (8-61757392-C-T) cells
HEK293 cells were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HEK293 intronic mutation (8-61757392-C-T) cells were transformation from HEK293 cells by introducing a intronic mutation and two synonymous mutations by homology-mediated end joining-based targeted integration using CRISPR/Cas9. The editor vector (addgene #48138) linked with sgRNA targeting sequence and the donor vetor homology with gene locus about 1.6kb exclude a intronic mutation and two synonymous mutations and flanked with sgRNA targeting sequence linked in pUC57 were liposome transfection by 1:1 ratio into HEK293 cells. Single GFP+ cells were isolated using flow cytometry and cultured. The intronic point mutation cells were identified by DNA sequencing. HEK293 synonymous control cells were similar to HEK293 intronic mutation cells with two synonymous mutations but not intronic mutation. All the HEK293 cells were cultured in DMEM (Gibco/Life Technologies) with 10% FBS (Gibco/Life Technologies) at 37 °C in 5% CO2 incubator (Thermo Scientific Heraeus).
Primers for cell genotypes as follows:
CHD7-forward: GAAGTTCACAGGAGCCAGAG
CHD7-reverse: CAGAAAGTAGAATGGTGATTGCCAG
Primary cultures of cortical neurons
Mouse cortical neurons were cultured from E14.5 C57BL/6 J of either sex. Cerebral cortices were dissected, dissociated, and cultured in 0.5 mL/well Neurobasal medium (Gibco, 21103-049) with 2% B27 (Gibco, 17504-044) and 2 mM Glutamax-I (Gibco, 35050-061) on Lab-Tek II Chamber Slide (Thermo Fisher Scientific, 154,941) at 100,000 cells/cm2. For the axon and dendrite experiment, the neurons were transfected by Lipo3000 (Invitrogen cat.L3000075) with totally 0.9 μg vector following LipofectamineTM 3000 Reagent Protocal 24 h after planting. After transfection, feed the cultures with new medium every 2 days.
Plasmid construct
The gene editor vector was sgRNA targeting sequence cloned in Cas9 vector (addgene #48138). The donor vector was homologous arm cloned in pUC57. The vector that expressed GFP was FUGW (addgene #14883). The control expression vector was GFP-removed FUGW. The CHD7 expressing vector was a gift from WeiJun Feng(Institutes of Biomedical Scienses Fudan University, Shanghai, China. The CHD7 (del and dup) expressing vector were modified by enzyme ligation and homologous recombination from CHD7 expressing vector. shRNA for mouse Chd7, human CHD7 and human TBR1 was cloned into FUGW-H1 vetor (addgene #25870). shRNA for control was DsRed.
shRNA sequence as follows:
shRNA for mouse Chd7: GCAGCAGCCTCGTTCGTTTAT
shRNA for human CHD7: GCAGCAGTCTCGTCCATTTAT
shRNA for human TBR1: GCCTTTCTCCTTCTATCATGC
shRNA for DsRed: AGTTCCAGTACGGCTCCAA
Whole-exome sequencing
DNA was extracted from peripheral blood of patients and their parents using the DNeasy Blood &Tissue Kit (Cat no. 69506, QIAGEN, GmBH, Germany), following the manufacturer’s instructions. For each sample to be sequenced, individual library preparations, hybridizations, and captures were performed following the protocol of Agilent SureSelect capture kit (V5) or IDT XGen Exome Research Panel. Sequencing was performed on an Illumina HiSeq X-10 instrument (Illumina) following the manufacturer’s protocol (HiSeq X-10 System User Guide).
Variation identify by Sanger sequencing
Based on the data from WES, all families within probands carrying CHD7 variations were selected for Sanger sequencing to validate whether variations are de novo or inherited from parents. The primers for Sanger sequencing as follows:
CHD7-forward: CCAGGGTTAGCTTTGTGGGT
CHD7-reverse: TGGCTTTGTGACCCTGTAGC
RNA isolation and reverse transcription
Each group of cells were dissociation in 1 mL Trizol (Invitrogen, 15,596,018). Total RNA was isolated using the method of user guide of TRIzolTM Reagent. Reverse transcribed using the Reverse Transcriptase M-MLV kit (TaKaRa, D2639B). 1 μg total mRNA and 50 nmol oligo dT as primer were used in the reverse transcription.
Quantitative real-time RT-PCR (qPCR)
For qPCR analysis, the gene expression of cDNA sample was analyzed using SYBR green (Toyobo, QPK-201). The qPCR program was three steps with melt as follows: 95 °C denaturation for 10 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 15 s, and 72 °C for 20 s. RNA level was calculated and standardized by using the ΔCt method and GAPDH expression level as control.
Primers for qPCR as follows:
CHD7 exon19-forward: ACGAAAAGGGGCCTATGGTG
CHD7 exon20-reverse: TTCAGCCTTCTTAGCCCACT
CHD7 exon25-forward: TCCCTGAACCTTTCCATGCT
CHD7 exon26-reverse: TCCCTGAACCTTTCCATGCT
CHD7 exon35-forward: ATGGCTGAAGCTGCACCCTA
CHD7 exon38-reverse: AGGCGGTCAAACATCGACTC
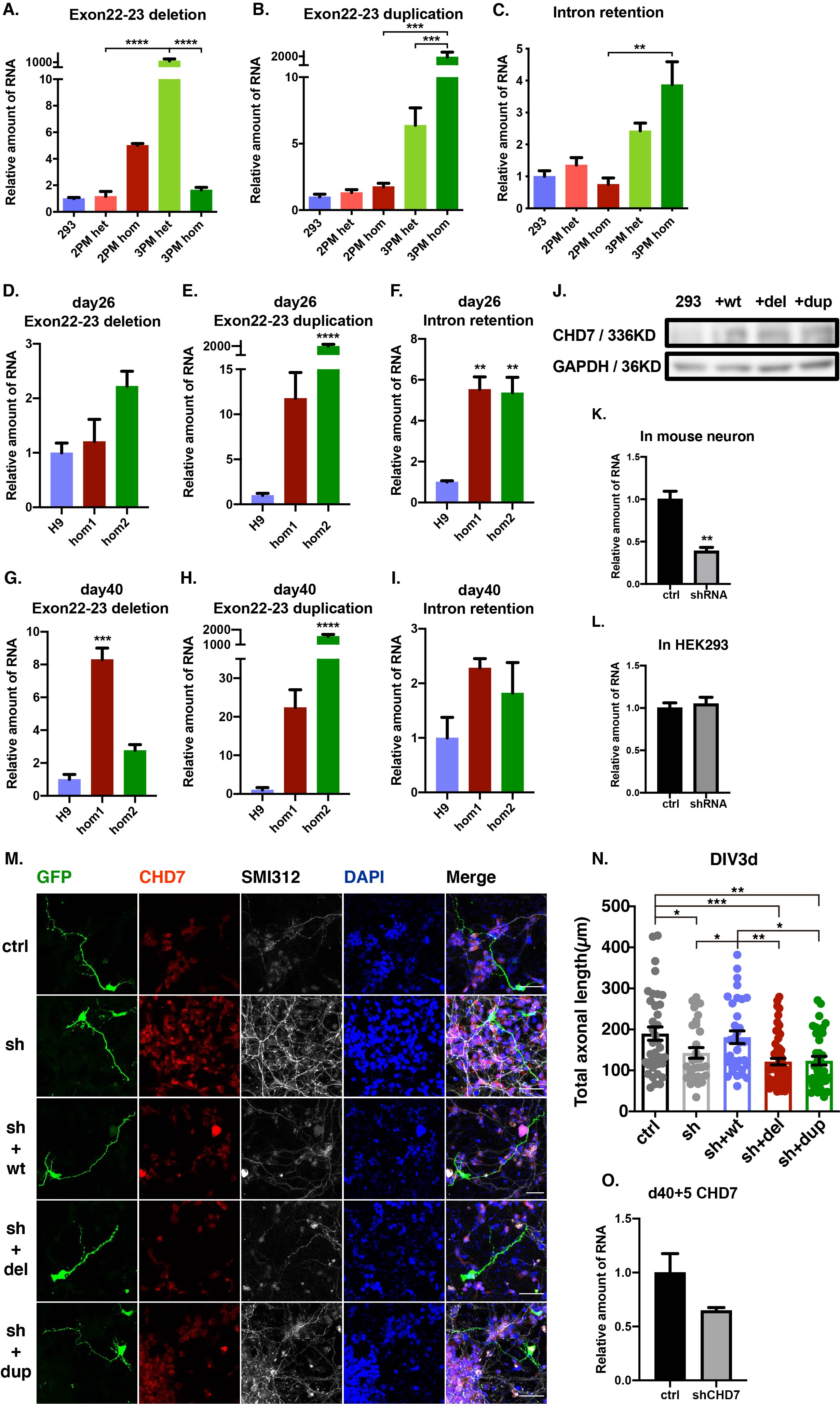
CHD7 intronic retention-forward: TGGAGAAGAATCTGCTTGTCTATGGG
CHD7 intronic retention-reverse: TCTGGGCTTTCACCTTCTTT
CHD7 exon22-23 deletion-forward: AATCTGCTTGTCTATGGGGTCC
CHD7 exon22-23 deletion-reverse: TCCCTGAACCTTTCCATGCT
CHD7 exon22-23 duplication-forward: CAACCATTCCGGTTTGTCAGC
CHD7 exon22-23 duplication-reverse: TCTGGGCTTTCACCTTCTTT
TBR1-forward: GACTCAGTTCATCGCCGTCA
TBR1-reverse: TGCTCACGAACTGGTCCTG
GAPDH-forward: CATCGCTCAGACACCATGGG
GAPDH-reverse: CCTTGACGGTGCCATGGAAT
Chd7-forward: TCCACATTTGCTAAGGCCAG
Chd7-reverse: TTCAGCCTTCTTAGCCCACT
Gapdh-forward: GTGAAGGTCGGTGTGAACGG
Gapdh-reverse: CGCTCCTGGAAGATGGTGAT
Differentiation of dorsal forebrain glutamate neurons
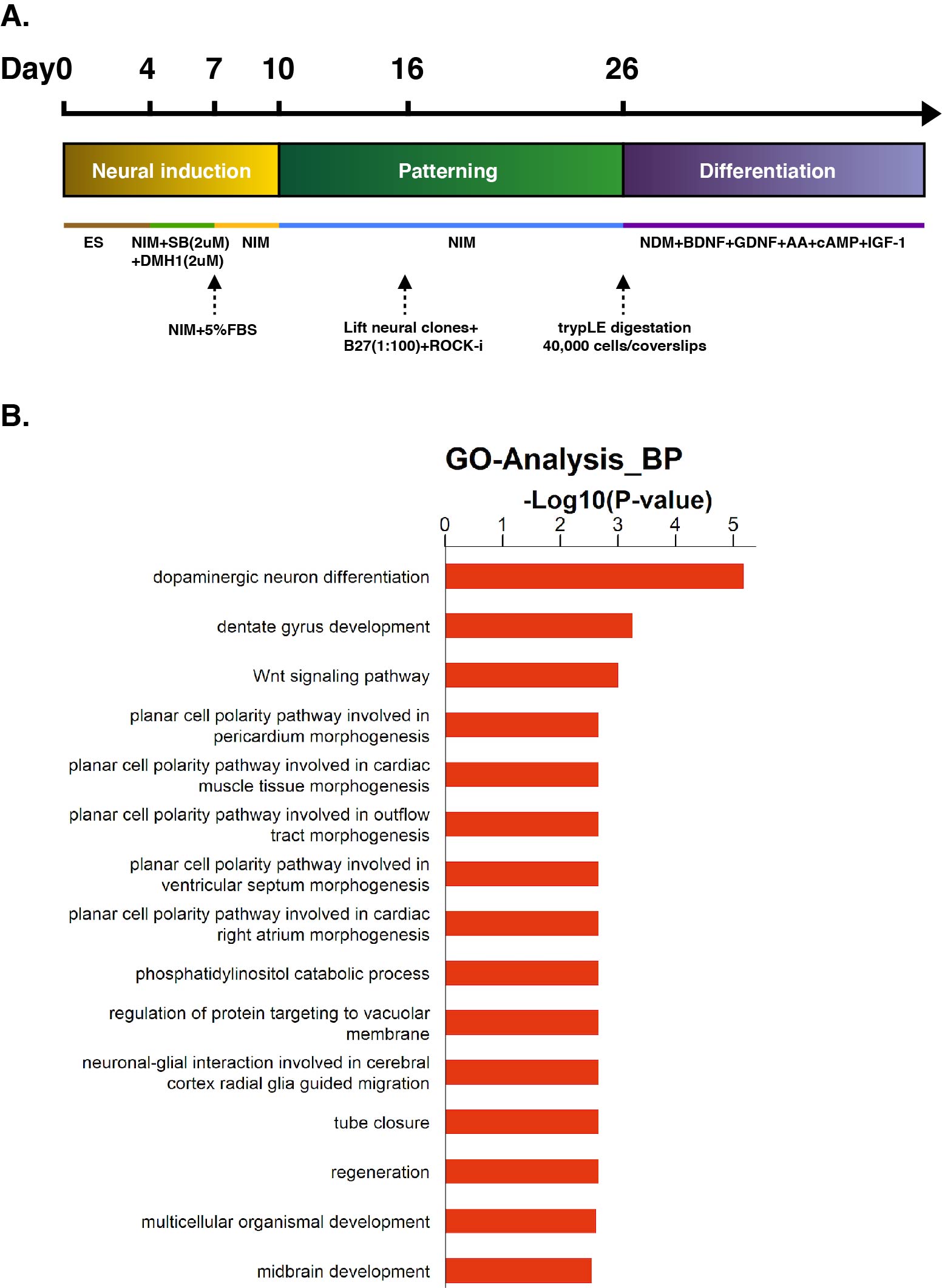
HESC colonies were cultured with daily medium change untile they reached about 80% confluence. Then, hESC clonies were detached from the feeder layer by digestion with dispase (Life Technologies), and then resuspended in hESC medium for 4 days to form embryoid bodies (EBs). For neural induction, EBs were cultured in neural induction media (DMEM/F12, 1% (v/v) N2 supplement, 5% (v/v) B27 without RA, 1% (v/v) NEAA, all from Life Technologies) (NIM) supplemented with SB-431542 (2uM, Stemgent) and DMH-1 (2uM, Tocris ) for 3 days. EBs were then attached to six-well plate in NIM supplemented with 5% fetal bovine serum. The cells were fed with NIM every other day till neural tube-like rosette formation at around day 16. Then, the rosettes were blown off by a 1-ml pipette and cultured in suspension. After two days, the cell clusters would form neurospheres, and then the medium was changed every other day. On day 26, neurospheres were digested into single cell by accutase, and seeded at a density of ~40,000 cells/cm2 on coverslips pre-coated with matrigel. After 5-6 hours, neuronal differentiation media (neural basal media, 1%(v/v) N2, BDNF (10ng/ml, PeproTech), GDNF (10ng/ml, PeproTech), cAMP (1uM, Sigma), IGF-I (10ng/ml, PeproTech) and AA (200uM, Sigma)) (NDM) was added to the wells, media was changed weekly.
Immunohistochemical staining
Cultured cells were washed with PBS for 5 mins, then fixed in 4% PFA at room temperature for 30 min. Wash every 10 mins with PBS twice. Block the cells with 5% BSA and 0.3% TritonX-100 in PBS at room temperature for 2 h. Incubate overnight at 4 °C with primary antibody in 3% BSA and 0.1% TritonX-100 in PBS. Wash with PBS every 10 mins for 3 times. Incubate at room temperature with secondary antibody and DAPI in PBS for 2 h. Wash with PBS every 10 mins for 3 times.
Primary antibody used and concentrations as follows:
Anti-SOX2 (R&D, AF2018, 1:500)
Anti-PAX6 (DSHB, AB-528427, 1:10)
Anti-Tuj1 (Sigma, T8660, 1:5000)
Anti-Ki67 (abcam, ab15580, 1:800)
Anti-CHD7 (CST, #6505, 1:1000)
Anti-GFP (abcam, ab6673, 1:400)
Anti-MAP2 (Millipore, MAB3418, 1:1000)
Anti-SMI312 (Biolegend, 837904, 1:1000)
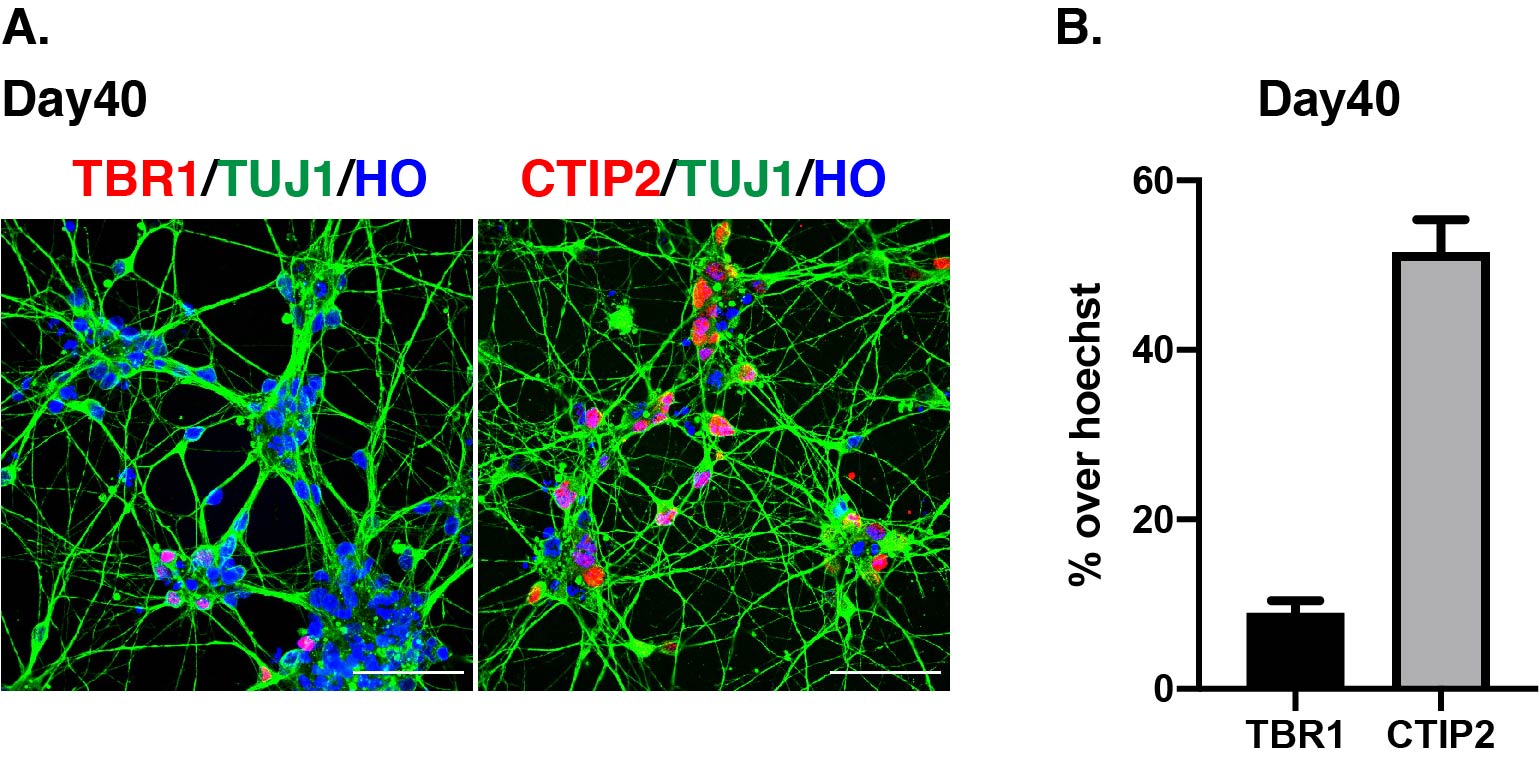
Anti-TBR1 (abcam, ab31940, 1:1000)
Anti-CTIP2 (abcam, ab18465, 1:200)
Hoechst (Life-tech/3570, 1:2000)
DAPI (Sigma, D9542, 1:1000)
Western blot
4-20% SDS-polyacrylamide gradient gel was used in the western blot. Electrophoresis program was 80 V 30mins and then 120 V 180 mins. Proteins were transferred onto the Immobilon polyvinylidene difluoride membrane (Millipore) for 210mins at 200 mA. The membrane was blocked by TBST solution (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) with 5% BSA for 2 h at room temperature and incubated overnight at 4 °C with primary antibody solution in 3% BSA. Wash with TBST every 10 mins for 3 times. The membrane was treated with secondary antibody for 2 h at room temperature. Wash with TBST every 10 mins for 3 times. The reaction was analyzed by using imaging film.
Primary antibody used and concentrations as follows:
Anti-CHD7 (CST, #6505, 1:1000)
Anti-GAPDH (ab8245, 1:5000)
Analysis of dendrites and axons
About 40–50 GFP-positive neurons were picked up randomly from each group. The searcher was blinded until statistical analysis was completed. The images were analyzed using Fiji software according to the standard: all of dendritic branches and secondary branch, the longest axon and secondary branch, and the total length of all neurites were taken into account. At least three independent experiments were performed,
Alternative splicing analysis
cDNA was segmental amplification by PCR. The products were separated by agarose gel electrophoresis and retrieved. Then ligated with pGEM-T Easy Vector. The ligation products were transformation into Escherichia coli Top10 and monoclonal culture. At least 40 monoclonal were sequenced per sanple.
Transcriptome analysis(RNAseq)
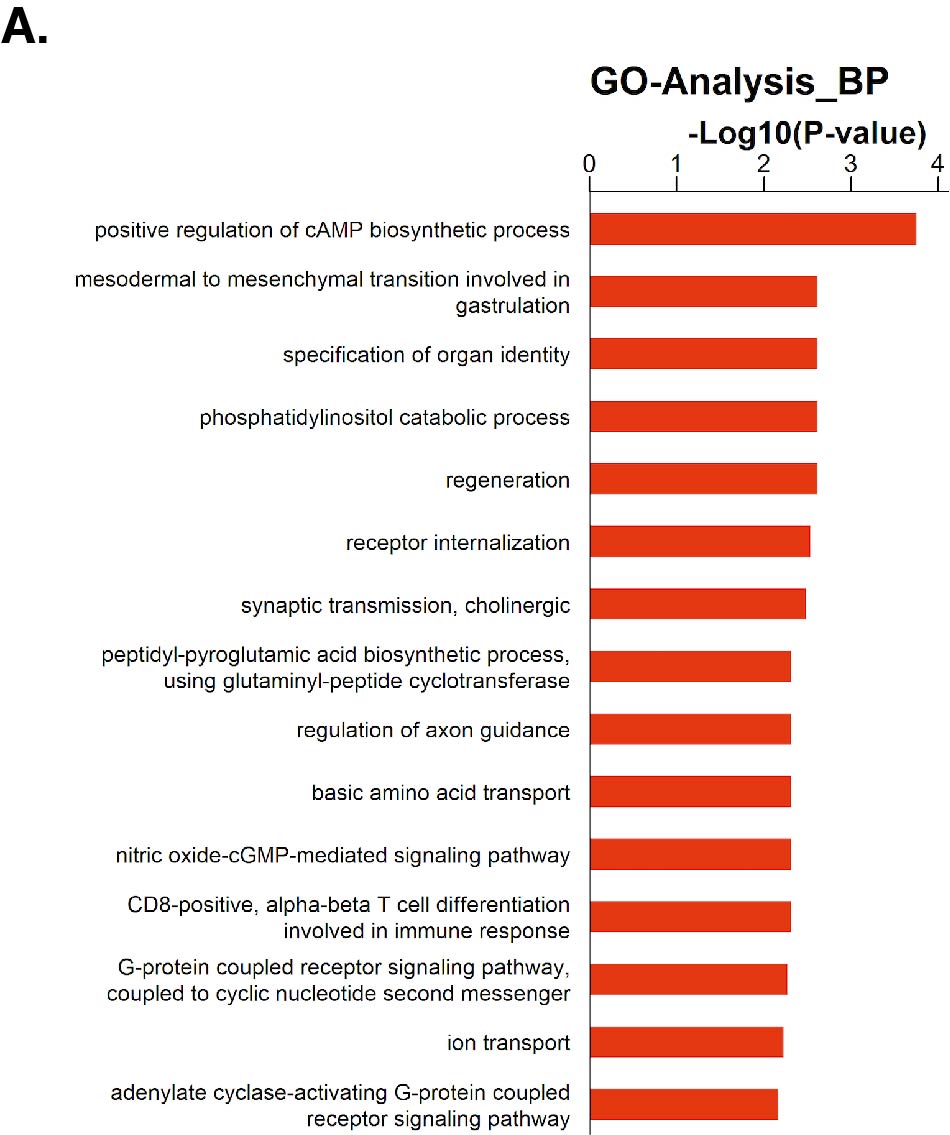
For RNA-sequencing, total RNA was extracted and subsequently a sequencing library was prepared by using the Illumila TrueSeq Total RNA Sample Prep Kit and sequenced on the Illumina Hi-Seq 2000. The clean reads were aligned to 9606(NCBI Taxonomy ID)genome (version: GRCh38) using the Hisat2. We applied HTseq to calculate the counts of the genes. RPKM/FPKM(Reads/Fragments Per Kilobase Million Reads)was used to standardize the expression data. We applied DEseq2 algorithm to filter the differentially expressed genes, then we filtered fold change and FDR under the following criteria: i) log2FC > 0.585 or < -0.585; ii) ,FDR < 0.05. For Gene ontology (GO) analysis, we downloaded the GO annotations from NCBI (http://www.ncbi.nlm.nih.gov/), UniProt (http://www.uniprot.org/) and the Gene Ontology (http://www.geneontology.org/). Fisher's exact test was applied to identify the significant GO categories and FDR was used to correct the p-values.
Statistical analysis
Statistical tests were carried out using GraphPad Prism 6 (Graphpad Software, lnc., RRID:SCR_002798). Two-tailed Student's t-test was used for sample pairs, one-way ANOVAs followed by Tukey's multiple comparison tests was used for 3 or more groups. Data distribution was test by Kolmogorov- Smirnov test and Shapiro-Wilk test by SPSS software (IBM, RRID:SCR_ 002865). The data distribution was normal distribution. Results were shown as mean ± SEM, and “n” represented to either the number of neurons (for morphological analysis) or the repeat experiment (for qPCR and RNAseq). Mouse cortical neurons were at least 3 times independently from 3 different litters. Stem cells differentiation were at least 3 batches independently. All data analyses were performed blinded to the experimental condition. All conditions statistically different from control are indicated. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Exclusion criteria: If the data was not in the 95% confidence interval of the group, the data would be excluded.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}