Study design and cohort summary
We recruited a cohort of 33 psoriasis subjects not on systemic therapy and 15 age- and gender- matched healthy individuals (Table 1a) to study the microbial features associated with psoriasis and their potential contribution to psoriasis pathogenesis. All psoriasis patients were clinically diagnosed with psoriasis at the UCSF Psoriasis and Skin Treatment Center and had a mean Psoriasis Area and Severity Index (PASI) of 14.2 representing moderate-to-severe disease. To characterize microbial composition, we collected stool samples and subjected each sample to shotgun metagenomic sequencing that provide both taxonomic composition and functional capacity. For the subsequent analyses, we focused on bacterial species, microbial UniRef90 gene families and microbial MetaCyc pathways. To link the changes of microbial features in psoriasis gut to the host response, we collected biopsies from sigmoid colon and subjected these samples to RNA-sEq. We also isolated PBMC from blood samples to measure immune cell profiles and cytokine production capacity. Together, our study design (Fig. 1) provided a comprehensive survey on both host biology and microbiome capacity in a cohort of psoriasis patients in comparison to healthy controls.
Microbial diversity and community structure between psoriasis and healthy gut microbiome
Gut microbiome dysbiosis has been previously associated with decreased microbial diversity. Low microbial diversity has been observed in several human diseases including IBD, obesity, and autism[11, 16, 17]. It has been hypothesized that losing microbial diversity rise from missing group of “beneficial microbes” in the gut microbiome, which can lead to many detrimental consequences such as loss control in growth of opportunistic pathogens and lack of production of beneficial microbial derived compounds. To compare the microbial diversity in psoriasis patients and healthy subjects, we calculated different alpha diversity indices to estimate overall diversity (Shannon), evenness (Simpson), and richness (chao1) of each community. We observed higher Shannon and Simpson diversity metrics in the gut microbiome of psoriasis patients driven by evenness but not richness of the microbial functional profile (Fig. 2A). In contrast, taxonomic diversity metrics at the species level are comparable between the psoriasis and healthy gut microbiome (Figure S1A). Despite increased functional diversity, the overall microbial community structures were similar between psoriasis samples and healthy samples as no distinct clusters was observed in PCoA plots for taxonomic and functional profile (Figure S1B and Figure S1C). All psoriasis patients in our cohort had a normal appearing lower gastrointestinal endoscopic exam, so major differences in diversity and community structure in the psoriasis microbiome as observed with other gastrointestinal diseases might not be expected. Transcriptomic profiles in colonic biopsies were comparable between psoriasis patients and healthy subjects. No gene features were significantly different between these groups after correction for multiple hypothesis testing (Figure S1D).
Identification of microbial features associated with the psoriasis gut microbiome
We hypothesized that even though the gut microbiome from psoriasis patients have seemingly normal overall microbial community structure and diversity, the differences between psoriasis and healthy microbiome may be in specific microbial features. To identify microbial features that are differentially abundant between gut microbiome from psoriasis patients and healthy individuals, we performed differentially abundance analysis using DEseq2[18] which estimates differential abundant features using negative binomial model after controlling for known confounding factors for gut microbiome such as gender, age and experimental batch. Our analysis revealed bacterial species and microbial gene families that are differentially abundant between microbiome in psoriasis patients and healthy individuals (Fig. 2C and 3A, Table S2 and S3). No microbial MetaCyc pathways were identified as differentially abundant. Among bacterial species with higher abundance in the psoriasis gut microbiome, Bacteroides vulgatus and Parasutterella excrementihominis (Fig. 2D) have been associated with other immune-mediated diseases. Increased intestinal colonization of Bacteroides vulgatus and elevated Bacteroides vulgatus reactive serum antibodies have been reported in ulcerative colitis patients[19]. Moreover, colonization of Bacteroides vulgatus is sufficient to promote colitis in several animal models, which further supports the active role of Bacteroides vulgatus in development of colitis[20–22]. Parasutterella excrementihominis belongs to a newly studied genera Parasutterella with limited literature. An increase in Parasutterella excrementihominis has been found in ileal submucosal tissue of Crohn’s disease patients [23] and in fecal samples of irritable bowel syndrome (IBS) patients [24]. Our analysis also revealed a decreased abundance of Phascolarctobacterium succinatutens in psoriasis compared to the controls (Fig. 2D). As suggested by its name, Phascolarctobacterium succinatutens utilizes succinate and converts it into acetate or propionate[25]. Accumulation of succinate has been observed in fecal samples of IBD patients[26, 27] and DSS induced colitis model in mice[28]. Moreover, reduced succinate utilizing Phascolarctobacterium was observed in both patients with Crohn’s disease and ulcerative colitis[14]. It is intriguing that the gut microbiome of psoriasis patients in our cohort shares some microbial features associated with IBD or IBS despite no intestinal symptoms reported in our patients. At the systemic level, we also detected a trend of elevated active CD4 + effector T cells in PBMC samples in psoriasis patients compared to healthy controls (Figure S2A), but no difference in cytokine production capacities (Figure S2B). Similar IBD-associated intestinal microbial signatures and elevated activated CD4 + effector T cells in the circulation of psoriasis patients suggest these patients may be more susceptible to systemic inflammation.
Microbial gene family analysis reveals three psoriasis subgroups with distinct microbial and host features
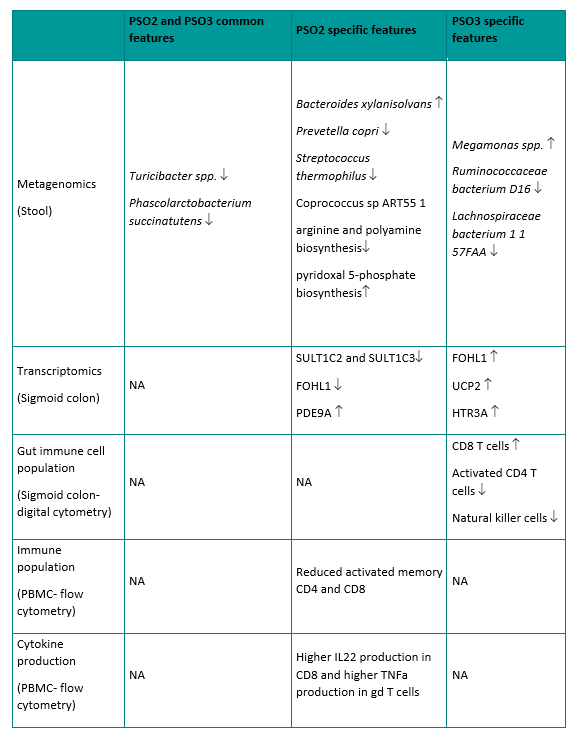
Although most microbial gene families that were differentially abundant between psoriasis patients and healthy controls had no functional annotations, hierarchical clustering analysis on these microbial gene families identified three distinct groups in our cohort (Fig. 3A and 3B). To confirm these subgroups were not observed by chance, we performed bootstrapped gap statistics on both the Euclidean distance and Bray-Curtis distance, which confirmed the presence of the three distinct groups in the cohort (Fig. 3C, Figure S3). Among the three groups identified by clustering, Group 1 consists of a mixture of healthy and psoriasis samples (14 healthy samples and 15 psoriasis samples), Group 2 consists of all psoriasis patients (9 psoriasis samples), and Group3 consists of almost all psoriasis patients except for 1 healthy control (1 healthy sample and 9 psoriasis samples) (Fig. 3D). The clustering was not confounded by BMI, age, and gender or diet (Figure S4A, S4B, S4C, S4G, Table S1). The clustering analysis revealed three distinct psoriasis subgroups, which highlight the heterogeneity of the disease. For the subsequent investigations of these psoriasis subgroups, we term the psoriasis subjects from these subgroups PSO1, PSO2, and PSO3. Specifically, psoriasis samples in group PSO1 clustered closely with most of healthy samples while group PSO2 and PSO3 represent more psoriasis dominant subgroups (Fig. 3A). All psoriasis subgroups had similar disease severity, disease duration, and age of disease onset (Figure S4D, S4E, and S4F). In addition, the microbial diversity at the level of species, gene families, and pathways were seemingly similar among the three psoriasis subgroups (Figure S4H, S4I, S4J). However, we found that each psoriasis subgroup had a number of distinct microbial and host features (Fig. 4A), which together suggest the biologic relevance of these psoriasis subgroups. Some of the interesting features associated with each psoriasis subgroup are highlighted below.
Interestingly, both PSO2 and PSO3 are dominated by psoriasis subjects, suggesting these are two distinct psoriasis specific subgroups. We identified several common microbial features shared by these psoriasis subgroups, as well as some microbial features that are distinct to each subgroup. While psoriasis samples have lower abundance in Phascolarctobacterium succinatutens compared to healthy controls (Fig. 2D), the reduced abundance of Phascolarctobacterium succinatutens is specific to PSO2 and PSO3, but not PSO1 (Fig. 4A). In addition, samples in PSO2 and PSO3 are less abundant with Turicibacter sanguinis and unclassified Turicibacter species (Fig. 4A). Less abundant Turicibacter species has been implicated in pediatric Crohn’s disease in several cohorts [29, 30]. Samples in PSO2 are more abundant with Bacteroides xylanisolvens and less abundant with Prevotella copri, Streptococcus thermophilus and Coprococcus sp ART55 1 (Fig. 4A). On the contrary, samples in PSO3 have lower abundance in Ruminococcaceae bacterium D16 and Lachnospiraceae bacterium 1 1 57FAA (Fig. 4A). Reduced abundance of Ruminococcaceae and Lachnospiraceae have been associated with the gut microbiome of psoriatic arthritis[6]. It is worth noting that all patients in PSO3 reported having joint pain or swelling, whereas only a fraction of patients in PSO1 or PSO2 did (Figure S4H). It is intriguing that the gut microbiome from patients in PSO2 and PSO3 share some microbial features observed in patients with IBD and psoriatic arthritis suggesting patients in these psoriasis subgroups may have increased association with these psoriasis comorbidities compared to patients in PSO1.
In addition to distinct taxonomic features, PSO2 has a distinct profile in microbial functions, especially in microbial pathways. Both abundant and depleted pathways were found in PSO2 relative to other psoriasis subgroups suggesting a shift of microbial functions in PSO2 (Fig. 4A). Microbial communities in PSO2 have lower abundance in the arginine and polyamine biosynthesis superfamily, suggesting lower capacity for polyamine production (Fig. 4A). Polyamines are small cationic molecules that modulate essential cellular process through binding of anionic DNA, RNA, and proteins. Polyamines are derived from arginine and intestinal polyamines can be generated by dietary supplementation, host metabolism, or microbial metabolism. Polyamines play a crucial role in intestinal mucosa maintenance and resident immune cell development [31]. Interestingly, it has been demonstrated that spermine, a class of polyamine, reduces IL-18 cytokine secretion by inhibiting NLRP6 inflammasome activation[32]. Samples in PSO2 also have increased capacity in pyridoxal 5-phosphate biosynthesis, which is a biologically active form of vitamin B6. Vitamin B6 can served as co-factor for various metabolic enzymes that are involved in wide range of cell metabolism pathways[33]. Intestinal commensal bacteria and dietary supplements are crucial source of vitamin B6 since mammalian cells lack the ability to de novo produce these micronutrients. Vitamin B6 deficiency has been linked to several immune-mediated diseases including rheumatoid arthritis and IBD[31, 34]. Our data suggest that gut microbial communities in PSO2 have increased microbe-derived vitamin B6 and reduced polyamine production (Fig. 4A). In contrast to PSO2, we did not identify microbial pathways that are uniquely associated with PSO1 and PSO3.
From the perspective of host response, PSO2 and PSO3 patients have distinct intestinal transcriptomic signatures (Fig. 4A). Gut biopsies from PSO2 patients have increased expression in ATP13A5 and PDE9A and reduced expression in sulfotransferases, SULT1C2 and SULT1C3 (Fig. 4A). In contrast, most of the transcriptomic signatures associated with PSO3 have increased expression compared to other psoriasis samples (Fig. 4A). It is interesting that some of the PSO3 transcriptomic signatures are similar to the ones reported in IBD patients or IBD animal models. FOLH1 encodes folate hydrolase, which is essential for absorption of dietary folate. Elevated FOLH1 expression has been reported in intestinal biopsies of patients with IBD and inhibiting FOLH1 activity ameliorates IBD associated abnormalities in mouse models [35, 36]. HTR3A encodes one of the serotonin receptors and increased HTR3A colonic expression has been observed in patients with Crohn’s disease [37]. UCP2 encodes for mitochondrial Uncoupling Protein 2 and has been implicated in several autoimmune diseases [38]. Expression of UCP2 is elevated in a DSS-induced mouse model of IBD and the severity of IBD can be ameliorated by knocking down UCP2 expression by siRNA[39]. Together, our data link PSO3 to IBD through intestinal transcriptomic profiles.
To gain more insight into the host intestinal immune response, we deconvoluted the immune cell composition in sigmoid colon bulk RNAseq using the digital cytometry framework CIBERSORTx[40]. As part of the CIBERSORTx framework, we first defined gene signatures of various immune cell populations in sigmoid colon using single cell RNAseq (scRNAseq) generated from healthy sigmoid colon[41]. We then used CIBERSORTx to apply the gene signature matrix to the bulk RNAseq to infer the composition of cell populations. Our analysis revealed differences in intestinal immune cell composition in different psoriasis subgroups which may represent distinct intestinal immune responses. Compared to other psoriasis subgroups, sigmoid colon from patients in PSO3 has more abundant CD8 + T cells and less abundant NK cells and activated CD4 + T cells (Fig. 4B). Although PSO3 patients possess some similarities in gene expression signature to IBD, PSO3 patients have less abundant colonic NK cells and activated CD4 + T cells, which are thought to be key contributors to IBD through cytokine production[42]. Interestingly, increased mucosal CD8 + T cell frequency has been reported in patients with Crohn’s disease[43] and CD8 + T cells can trigger intestinal inflammation in mice colitis model[44]. Our data suggest PSO3 patients may be linked to IBD and particularly Crohn’s disease through a CD8 + T cell mediated pathogenesis.
In addition to transcriptomic signatures, we observed distinct features in circulatory immune profiles and in the gut transcriptome between the two psoriasis dominant groups. Patients in PSO3 have higher activated memory CD4 + effector T cells compared to PSO2 while the frequency of memory CD4 + effector T cells and total CD4 + effector T cells are comparable between these two psoriasis subgroups (Fig. 4C). Similarly, memory CD8 + T cells have more activated population in PSO3 patients (Fig. 4C). Despite low T cell activation, patients in PSO2 have a higher capacity to produce IL-22 by circulating CD8 + T cells and TNF-alpha by circulating gamma-delta T cells (Fig. 4C) compared to healthy controls or PSO1. Together, our data reveal differential underlying circulatory immune responses associated with PSO2 and PSO3. PSO2 patients have higher capacity for pro-inflammatory cytokine production, while PSO3 patients have higher baseline T cell activation.
Distinct correlations between psoriasis severity to microbial features in each psoriasis subgroups
To further understand the relationship between the psoriasis subgroups and psoriasis heterogeneity, we tested if microbial features in these psoriasis subgroups are significantly correlated with psoriasis parameters such as severity, disease onset, and duration. We detected no significant correlations between the microbial features and any of the disease information when we combined all psoriasis patients in the cohort (data not shown). However, the microbial features-disease parameter correlations were observed in individual psoriasis subgroups. In PSO1 patients, disease severity is correlated with microbial pathways involved in purine degradation (Fig. 4D). Several microbial pathways are positively correlated with disease severity in PSO2 patients including pentose phosphate biosynthesis and L-arginine biosynthesis (Fig. 4D). The microbial L-rhamnose degradation pathway is positively correlated with disease duration in PSO2 (Fig. 4D). The microbial L-isoleucine biosynthesis pathway is correlated with disease duration positively in PSO1 patients and negatively in PSO2 patients (Fig. 4D). No significant correlations between microbial features and disease information were observed in PSO3. The different correlations in psoriasis subgroups might reflect the differential impact of gut microbiome in modulating psoriasis progression.
Multi-omic analysis in psoriasis patients and subgroups reveals distinct host-microbe interactions
In order to gain a comprehensive view of the crosstalk between the gut microbiome and host biology, we constructed multi-omic correlation networks by integrating microbial and host features across different measurements (Table S9). A subset of 40 subjects from our cohort with complete measurements from shotgun metagenomic sequencing, gut RNAseq, and circulating immune profiling were included in the multi-omic analysis. This multi-omic cohort consists of 26 psoriasis subjects and 14 age- and gender-matched healthy subjects (Table 1b). Multi-omic networks were constructed within each disease status and within each psoriasis subgroup to reveal disease specific and subgroup specific host-microbe relationships. We applied Spearman’s rank correlations to features from different measurement types and identified strong and robust correlations using the following criteria: (1) the correlations had adjusted p-value smaller than 0.1, (2) the correlations had absolute correlation coefficient greater than 0.6, (3) the correlations had features with at least 70% non-zero counts to avoid correlations that are driven by few extreme data points. The resulting multi-omic networks consist of nodes that represent host or microbial features and edges that represent significant correlations between nodes. Compared to the multi-omic network in healthy subjects, the network in psoriasis patients are denser and consists more edges (Fig. 5A). The final multi-omic network consists of 73 significant correlations within healthy subjects and 97 significant correlations within psoriasis patients (Table 2). Intriguingly, the multi-omic network in each psoriasis subgroup displayed a distinct network structure. Multi-omic networks in PSO1 and PSO2 were more interconnected compared to the network in PSO3 (Fig. 5A). It is interesting that PSO3 had fewer edges and nodes compared to other psoriasis subgroups and all psoriasis samples combined (Table 2). Overall, this analysis revealed distinct multi-omic network structures in psoriasis patients and healthy controls, as well as among different psoriasis subgroups.
The different multi-omic networks might reflect the different host-microbe interactions in psoriasis and healthy subjects. We hypothesize that the unique host-microbe interactions might contribute to pathogenesis of psoriasis and its comorbidities. Indeed, we identified inflammation associated modules in the multi-omic network of psoriasis patients. One of the modules in the psoriasis associated multi-omic network positively links microbial pathways involved in microbial ppGpp biosynthesis and pyrimidine ribonucleosides salvage pathways with circulating IL-17 production in both CD4 + effector T cells and regulatory T cells (Fig. 5B). IL-17 cytokines are key disease drivers in psoriasis and blocking IL-17 can effectively ameliorate inflammation in psoriatic skin[45]. The associations between these microbial pathways and IL-17 production were only observed in psoriasis samples despite these microbial pathways are also being abundant in healthy samples (Figure S5A). Our analysis also revealed psoriasis specific associations between colonic expression of two pro-inflammatory chemokines, CXCL1 and CXLC3, and three unannotated microbial gene families (Fig. 5C, Figure S5B). Increased expression of CXCL1 and CXCL3 is associated with an inflammatory state of the intestine [46]. Our result suggests potential microbial controls of colonic expression of CXLC1 and CXCL3 in psoriasis patients prior to developing intestinal inflammation.
Besides psoriasis specific host-microbe interactions, our analysis also identified host-microbe interactions that are specific in psoriasis subgroups. A multi-omic network identified in PSO1 consists of a module that associates microbial gene families with activated non-memory CD4 + T cells and IL-17 production capacity in CD4 + effector T cells (Fig. 5D), suggesting potential microbial controls in T cell activation and effector function in this psoriasis subgroup. Indeed, 95 out of 124 host-microbe associations in PSO1 link features of host circulating immunity to microbial features, which is higher compared to networks in PSO2 and PSO3 patients (Table 2). In PSO2, we identified many associations linking microbial features to colonic transcriptome (Fig. 5A) including genes associated with IBD. CXCL8/IL8 is a chemokine that attracts neutrophils to the site of inflammation and elevated expression of CXCL8/IL8 has been found in inflamed ulcerative colitis sigmoid colon [47]. Our data show that the expression of CXCL8 is positively correlated with microbial putrescine biosynthesis pathway in PSO2 patients (Fig. 5E). In addition, microbial sugar metabolism pathways are negatively correlated with colonic expression of FOS and PDE4C (Fig. 5F). FOS encodes c-fos which is a key subunit of AP-1, which is an important transcriptional factor regulates immune function. It has been shown that c-fos plays a protective role in suppressing inflammation in both LPS and DSS induced colitis models[48, 49]. PDE4C belongs to PDE4 family, which is a family of phosphodiesterase that degrades cyclic AMP (cAMP), which is an anti-inflammatory signaling molecule. Inhibiting PDE4 activity has been demonstrated as an effective strategy to treat inflammatory disease including psoriasis and IBD [50]. Together, our results suggest potential microbial controls of host colonic immune response through microbial metabolism in PSO2 patients. Compared to multi-omic networks in PSO1 and PSO2, PSO3 associated network has fewer associations (Fig. 5A, Table 2). Despite the sparse network, we identified a module in PSO3 network negatively correlates microbial tetrapyrrole biosynthesis pathway with several genes in B-cell biology including CD79B, PAX5, TCL1A, and MYBL1 (Fig. 5G). Tetrapyrroles are metal binding compounds consist of four pyrrole rings. With different modifications and metal bound, tetrapyrroles serve as different cofactors, such as heme, cobalamin (Vitamin B12), and coenzyme F430 and have crucial roles in regulating diverse cell functions. The role of tetrapyrroles in B-cell functions has not been described. However, IBD patients are found to have depleted tetrapyrroles in their fecal samples [51] and increased colonic humoral immune response [52]. The negative association between microbial tetrapyrrole pathways and B cell genes found in PSO3 patients reminiscent of changes in tetrapyrroles level and humoral immune response observed in IBD patients may suggest an increased risk of developing IBD for PSO3 patients.