Animals
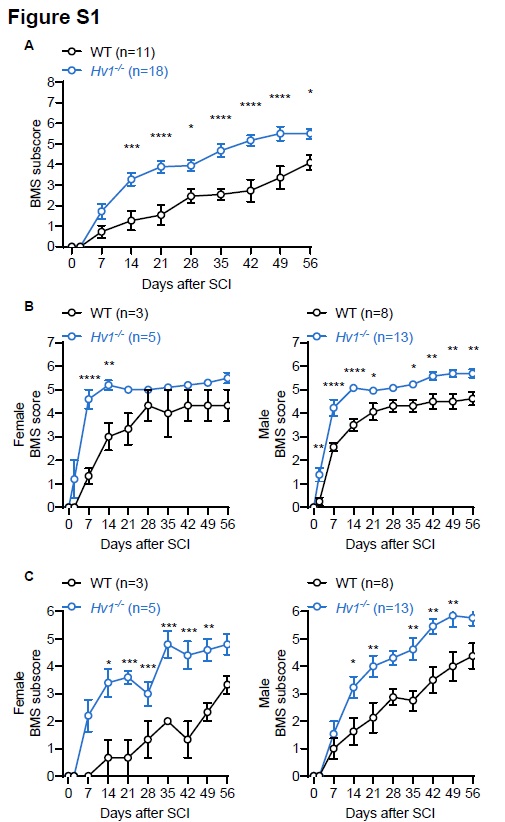
All experiments were conducted using adult male C57BL/6J mice or Hv1−/− mice (20–30 g). Female mice were only used to investigate the role of sex difference in locomotor recovery. All mice were housed on a 12:12hlight/dark cycle with food and water available ad libitum. The experiements were performed in accordance with institutional guidelines, as approved by the animal care and use committee at Rutgers University and Mayo Clinic. The animals were monitored on a daily basis after SCI. Any unintended changes in appearance (disheveled hair, weight loss, and dehydration), behavior (decreased grooming, eating and drinking) and activity (decreased exploring and nesting) were noted and euthanized if no improvement was observed.
Sci Contusion Model And Post-surgery Animal Care
Mice were anesthetized with ketamine (60 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and a laminectomy over the dorsal portion of T9. The spinal column was stabilized via the lateral processes using clamps at T8 and T13 vertebraes. The exposed dorsal surface of the spinal cord was subjected to a 3-g weightdrop with tip diameter of 0.5-mm flat surface, modified NYU impactor, from a height of 6.25 mm using a modified NYU impactor resulting in a moderate contusion injury [43]. After the injury, a small piece of subcutaneous fat was placed on the dorsal surface before the skin was closed, in order to avoid tissue adhesion between dura and peripheral tissues. For sham control, laminectomy was performed without injuring spinal cord. After SCI, mice were subcutaneously injected with 0.1 ml 0.125% (50 µl per 25 g body weight of mice) bupivacaine near the surgical area 5 min after surgery for post-operative analgesia. 0.5 ml Saline (SAL-090; Rocky Mountain Biologicals) and 0.05 ml 20 mg/ml cefazolin (054846; Henry Schein) were subcutaneously injected daily for 3 days. Mice were kept on absorbent bedding and their bladders were manually expressed twice each day until a reflex bladder was established (14–28 d after SCI).
Locomotion Recorvery Assessment
Mice were tested for locomotor recovery of the hindlimbs in an open-field chamber on day 2 after injury and weekly thereafter for up to 8 weeks using the Basso mouse scale (BMS) for locomotion [44]. Successful contusion surgery resulted in complete motor function loss of the hind limbs 2 h after surgery impact (BMS score 0, no ankle movement), whereas successful sham surgery models had no effect on hind limb mobility (BMS score 9, frequent or consistent plantar stepping, mostly coordinated, paws parallel at initial contact and lift off, normal trunk stability, and tail up).
Whole-cell Patch Clamp Recording
Live spinal cord slices were prepared at 7d after SCI in WT (CX3CR1GFP/+ mice) and Hv1−/− (Hv1−/− CX3CR1GFP/+) mice or in sham controls [30, 45]. Briefly, mice were anesthetized in isoflurane and perfused with ice-cold artificial cerebrospinal fluid (ACSF). The spinal cord was swiftly removed and the slices (300 µm) were made using a vibratome in ice-cold oxygenated (95% O2 and 5% CO2) ACSF with the following composition (in mM): NaCl, 124; NaHCO3, 25; KCl, 2.5; KH2PO4, 1; CaCl2, 2; MgSO4, 2; glucose, 10 and sucrose added to make 300-320mOsmol. The slices were then transferred to a recovery chamber for 30 m with oxygenated ACSF with the same composition as above at room temperature before electrophysiological studies. Whole cell patch-clamp recordings were made on microglia with CX3CR1GFP/+ transgencially labeled (~ 50 µm deep). Recording electrodes (4 − 5 MΩ) contained a K-based internal solution composed of (in mM): 120 K-gluconate, 5 NaCl, 1 MgCl2, 0.5 EGTA, 10 Na2Phosphocreatine, and 10 HEPES (pH 7.2; 280–300 mOsmol). The membrane potential was held at -20 mV. Data were amplified and filtered at 2 kHz by a patch-clamp amplifier (Multiclamp 700B), digitalized (DIGIDATA 1440A), stored, and analyzed by pCLAMP (Molecular Devices, Union City, CA). Data were discarded when the input resistance changed > 20% during recording. The voltage ramp test was performed from − 100 to 100 mV in 500 ms. For electrophysiology, a minimum of five cells from at least three different mice from the same litter were randomly selected for recording per condition.
Tissue Preparation And Cryosection
Mice were deeply anaesthetized by isoflurane and perfused transcardially with 40 ml of cold phosphate-buffered saline (PBS) followed by 40 ml of cold 4% paraformaldehyde (PFA) in PBS [46]. The spinal cord was removed and post-fixed with the same 4% PFA for 6 h at 4 °C, then transferred to 30% sucrose in PBS for 48 h. Tissues were embeded in optimal cutting temperature compound (OCT compound) prior to frozen sectioning. Cryosection was performed to collect complete 2–4 mm region containing the injuried epicenter. Sample sections (15 mm in thickness) were prepared on gelatin-coated glass slide with a cryostat (Leica) for further staining.
White Matter Sparing Analysis
Hematoxylin-eosin (H&E) staining was used to examine white matter sparing after SCI. Thermo Scientific™ Shandon™ Rapid-Chrome H & E Frozen Section Staining Kit was applied on spinal cord frozon sections according to manufacturer‘s instructions (9990001, Thermo Scientific™). Frozen spinal cord sections mounted on glass slides were allowed to reaclimate to room temperature for at least 30 min, and then washed with (tris buffered saline )TBS for 10 min. According to staining protocol, the slides were placed in Rapid-Fixx solution for 5–7 s. Then slides were dipped into distilled water for 10 times. After that, slides were incubated in hematoxylin for 1 min, dipped into distilled water for another 10 times, and dipped 3 times in bluing reagent. Enriched positive charges allowed Hematoxylin to bind DNAs, and turned nucleus into blue by bluing reagent. Slices were then dehydrated in 95% alcohol by 5–7 dips, followed by 15 s Eoxin-Y solution, which stained cytoplasm with red or pink. Finally, slices were dipped 5–7 times into 95% alcohol, 100% alcohol, 100% alcohol again, xylene, and xylene again, in turn, for the purpose of dehydration and clearing. The slides were imaged with a bright-field microscope. Grey matter had more densed cells (nucleus) compared to the white matter, thus H&E staining enhanced the contrast between grey and white matter. The perimeter of the white matter was traced and the enclosing area was measured using ImageJ software (National Institutes of Health, Bethesda, MD).
Immunofluorescence Staining
Frozen spinal cord sections mounted on glass slides were allowed to reaclimate to room temperature for at least 30 min, and then washed with TBS to remove OCT compound. The slices were blocked with 5% goat serum in 0.4% Triton X-100 in TBS for 60 min, and then incubated overnight at 4 °C with the following primary antibodies: rabbit-anti-Iba1 antibody (1:500; 019-19741; Wako, Japan); rabbit anti-NeuN (1:1000; ab104225; Abcam, USA); mouse anti-IL-1β (1:300; 12242; Cell Signaling, USA). After primary antibody incubation, slides were re-warmed to room temperature for 30 min and washed with TBS three times for 15 min each wash. Then slices were incubated for 90 min at room temperature with secondary antibody in the dark (1:500, Alexa Fluor 594 goat anti-rabbit, Alexa Fluor 488 goat anti-mouse, Life Technologies). The sections were mounted with DAPI Fluoromount-G (SouthernBiotech) for further imaging.
Quantitative Image Analysis
Fluorescent images were obtained with a fluorescence microscope (EVOS FL Cell Imaging System, Life Technologies). Cell counting, soma size analysis and fluorescent signal intensity were quantified using ImageJ software (National Institutes of Health, Bethesda, MD). For the quantification of microglia and macrophages (Iba1+) and neurons (NeuN+), the total number of immunolabeled cells stained with nuclear marker DAPI was counted at 20x magnification images. For the quantification of IL-1β, imaging parameters were kept constant to avoid technical artefacts in the analysis of fluorescent signal intensity.
Reactive Oxygen Species Assay
ROS content in spinal tissue homogenates were measured using the OxiSelect™ ROS/RNS assay kit (Cell Biolabs, STA- 347) according to the manufacturer‘s instructions. In brief, mice were perfused transcardially with 40 ml cold PBS. Approximately 16 mg fresh spinal cord samples containing the SCI epicenter were extracted and homogenized with tissue extraction reagent Ⅰ (10 ml PER 1 g of tissue; Invitrogen FNN0071) with protease and phosphatase inhibitors, and then supernatant was collected after centrifugation. Lysate was added to the wells along with, dichlorodihydrofluorefscin (DCF), a ROS specific flurorescent probe in the presence of a catalyst. The resultant fluroescent intensity was proportional to the amount of free radicals in the sample. The assay was performed in a 96-well plate format using a standard fluorescence plate reader. The free radical content in samples was determined by comparison with the predetermined DCF standard curve.
Cytokine Array
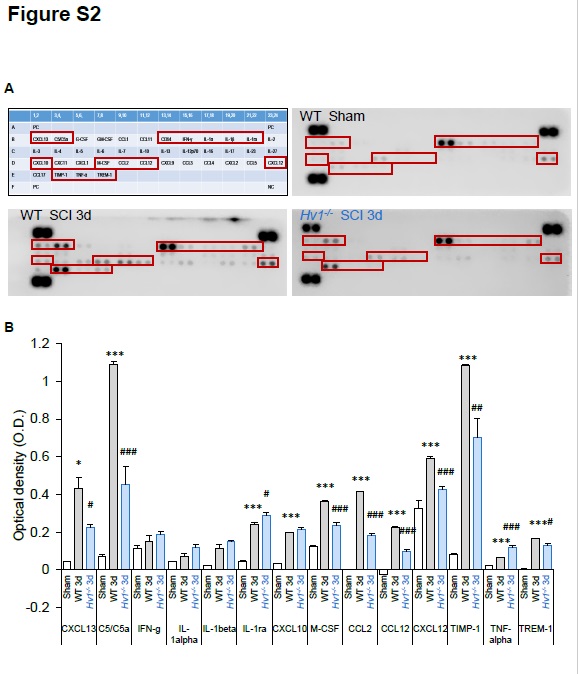
Proteome Profiler Mouse Cytokine Array Kit (R&D stystems, ARY006) was applied to assess the inflammatory response according to manufacturer’s instructions [47]. In brief, mice were anesthetized by isoflurane and perfused transcardially with ice-cold PBS. Protein samples were prepared in the same way as for ROS assay and 300 µg of proteins collected from the spinal cord was used for each sample and were run as duplicates.
Statistical Analysis
All data are presented as the mean ± SEM and were analyzed for significant differences between groups by two-way ANOVA with post hoc Tukey test for multiple comparisons. In all cases, p < 0.05 was considered to be significant.
{kind=link}
{kind=link}