Ethics statement
Healthy human deciduous teeth were collected as discarded biological/clinical samples from three healthy donors (5–7 years old) who attended the Department of Pediatric Dentistry of Kyushu University Hospital. The parents/guardians of the donors provided written informed consent that stem cells harvested from the teeth could be used for research into regenerative medicine for congenital diseases. Human peripheral blood mononuclear cells (PBMNCs) were collected from peripheral venous blood obtained from donors (25–28 years old) unrelated to those who provided deciduous teeth. Procedures for handling human samples were approved by Kyushu University Institutional Review Board for Human Genome/Gene Research (Protocol Number: 393-00).
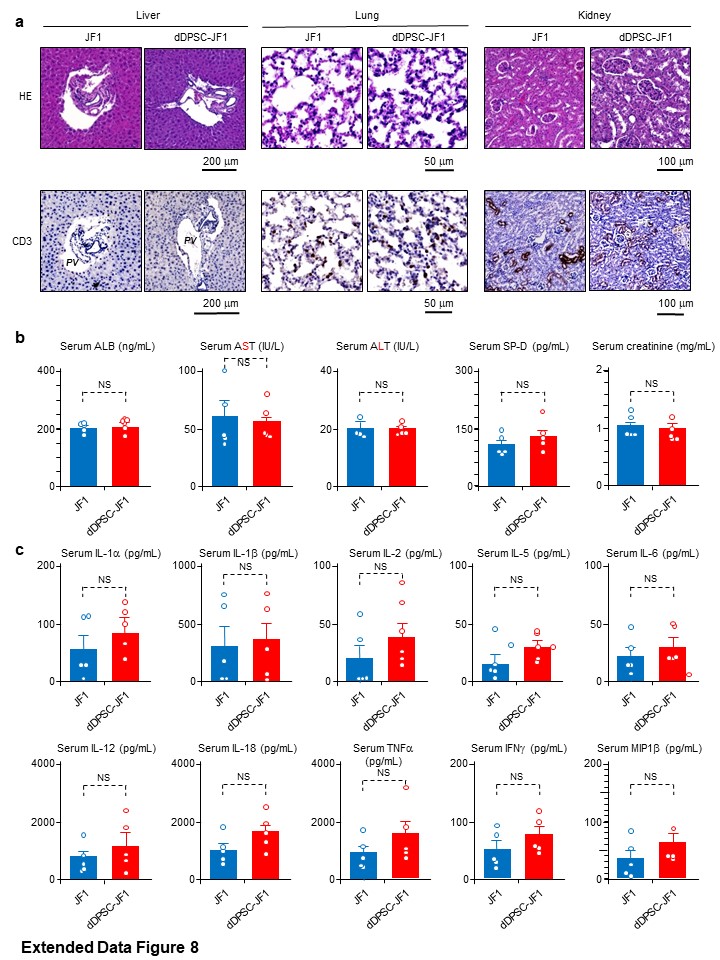
All animal experiments were approved by the Institutional Animal Care and Use Committee of Kyushu University (Protocol Number: A21-044-1; A25-086-0) and Nagoya University (Permissions #28316, #29121, #30312, #31199 and #20430). Stem cell transplantation caused no substantial biological alterations or welfare issues in the animals (Fig. 4, Extended Data Fig. 8). All experiments were performed in accordance with relevant guidelines and regulations.
Animals
Male JF1 mice (National Institute of Genetics, Mishima, Japan) and male and female wild-type C57BL/6J (B6) mice (Kyudo, Tosu, Japan; Japan SLC, Hamamatsu, Japan) were housed under controlled environmental conditions (12-h light/12-h dark cycle) and given free access to sterile drinking water and a standard MF chow diet (Oriental Yeast, Tokyo, Japan).
Stem cell isolation and culture
Isolation and culture of stem cells from dental pulp tissue were performed using enzyme digestion and colony-forming unit fibroblast (CFU-F) methods14,15,42,43. Remnant dental pulp tissues from extracted human deciduous teeth were digested with 0.3% collagenase type I (Worthington Biochemicals, Lakewood, NJ, USA) and 0.4% dispase II (Sanko Junyaku Co., Ltd., Tokyo, Japan) for 60 min at 37ºC. The resultant cells were passed through a 70-µm cell strainer and seeded on T-75 culture flasks. Three hours after cell seeding, the culture flasks were washed with sterilized Ca2+-free and Mg2+-free phosphate-buffered saline (PBS). The remaining adherent cells were incubated in Minimum Essential Medium Eagle-Alpha Modification (αMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 15% foetal bovine serum (FBS; Equitech-Bio, Kerrville, TX, USA), 100 µM L-ascorbic acid 2-phosphate (Wako Pure Chemicals, Osaka, Japan), 2 mM L-glutamine (Nacalai Tesque, Kyoto, Japan) and premixed 100 U/mL penicillin/100 µg/mL streptomycin (Nacalai Tesque) for 16 days. The resulting colonies were passaged to expand the number of dDPSCs. Cells at P3 were used for the experiments.
BMMSCs isolated from human whole bone marrow aspirates obtained from healthy adult volunteers (n = 3; AllCells, Alameda, CA, USA) and human skin fibroblasts (FBs; PromoCell, Heidelberg, Germany) were prepared as described previously15.
Stem cell characterization
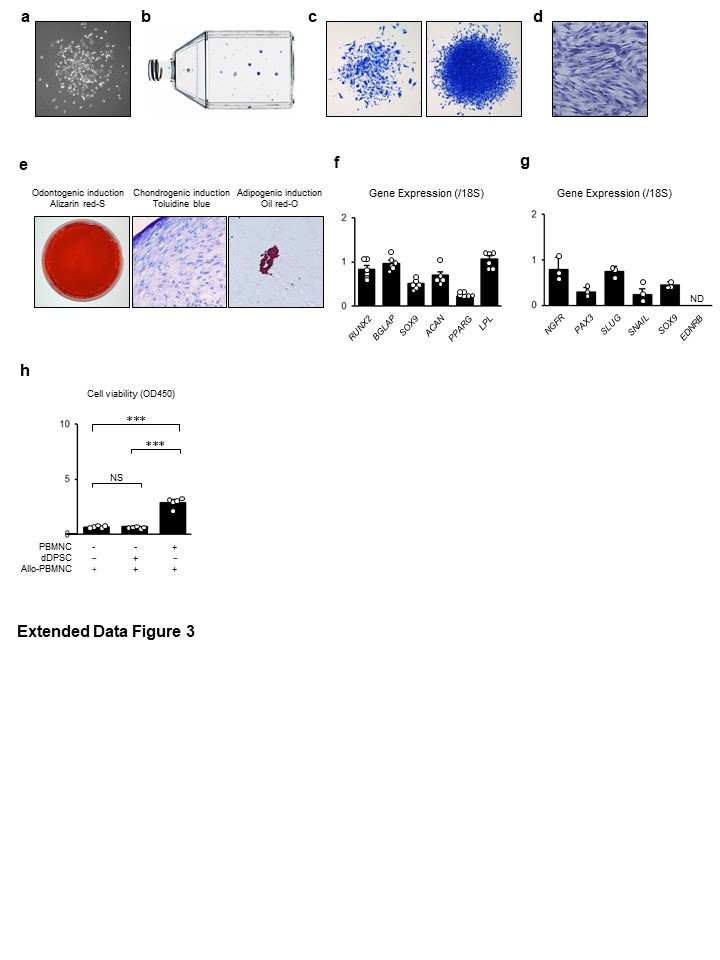
dDPSCs were characterized according to standard protocols44. The adherent colony-forming capacity was evaluated using a CFU-F assay. Isolated cells (10 ´ 103) were seeded on T-75 culture flasks and cultured in growth medium for 16 days. The flasks were treated with 4% paraformaldehyde and 0.1% toluidine blue in PBS (pH 7.4) for 18 hours. The number of fibroblast colonies containing >50 cells was counted under a microscope.
The multipotency of dDPSCs (P3) was verified according to previous studies12,15. Briefly, specific culture conditions were used to induce dDPSCs to differentiate into osteoblasts, chondrocytes or adipocytes that were stained with alizarin red-S, toluidine blue or oil red-O, respectively. Expressions of genes specific to osteoblasts (runt-related transcription factor-2 [RUNX2] and osteocalcin [BGLAP]), chondrocytes (SRY-box-9 [SOX9] and aggrecan [ACAN]) and adipocytes (peroxisome proliferator-activated receptor gamma-2 [PPARG] and lipoprotein lipase [LPL]) were analysed by quantitative reverse transcription polymerase chain reaction (RT-qPCR). Expressions of genes specific for neural crest cells, including nerve growth factor receptor (NGFR), paired box-3 (PAX3), zinc finger protein SNAI-2 (SLUG), zinc finger protein SNAI-1 (SNAIL), SOX9 and endothelin receptor type B (EDNRB), in dDPSCs (P3) were examined by RT-qPCR. Expressions of CXCR4 and KIT in dDPSCs (P3) were assessed using immunofluorescence experiments (see Supplementary Table 1). We have previously confirmed that dDPSCs cultured under the same conditions express stem cell markers (e.g., CD146) and low levels of class II HLA and T cell costimulatory molecules15.
For mixed lymphocyte assays, human PBMNCs (1 × 106) were co-cultured with dDPSCs (100 × 103) and gamma-irradiated human PBMNCs (100 × 103; 30 Gy irradiation with an MBR-1520R-3 irradiator; Hitachi, Tokyo, Japan) or stimulated with phytohemagglutinin (5 μg/mL; Merck, Darmstadt, Germany) in RPMI-1640 medium (Sigma-Aldrich) containing 10% heat-inactivated FBS (Equitech-Bio), 2 mM L-glutamine, 1 mM sodium pyruvate and premixed 100 U/mL penicillin/100 µg/mL streptomycin (Nacalai Tesque). Five days after co-culture or stimulation, cell viability was assayed using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) and a Multiscan GO plate reader (Thermo Fisher Scientific).
Stem cell transplantation
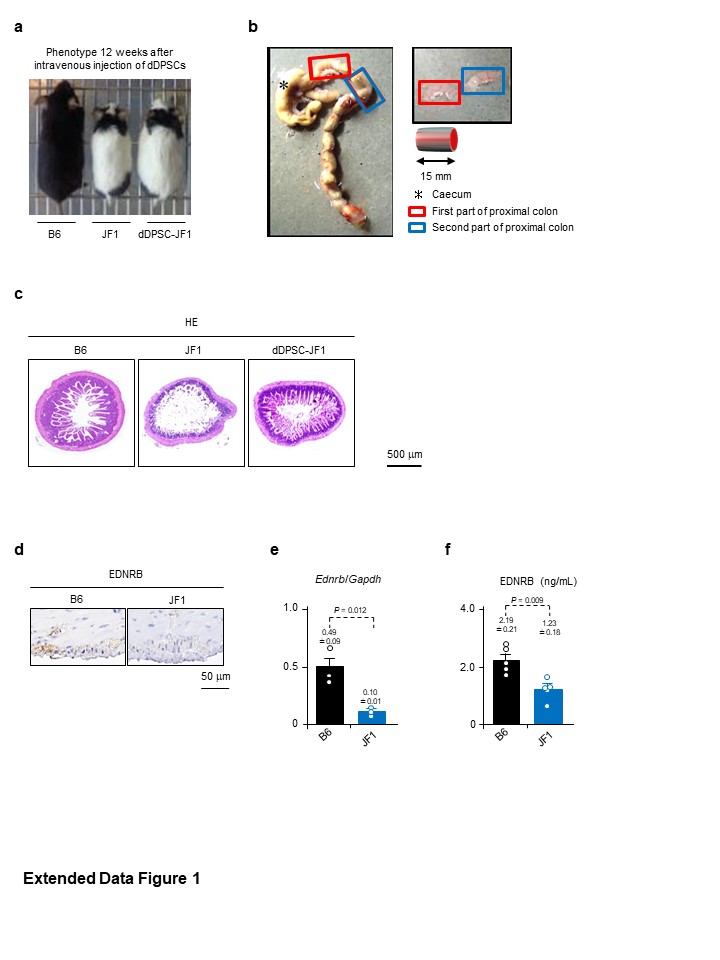
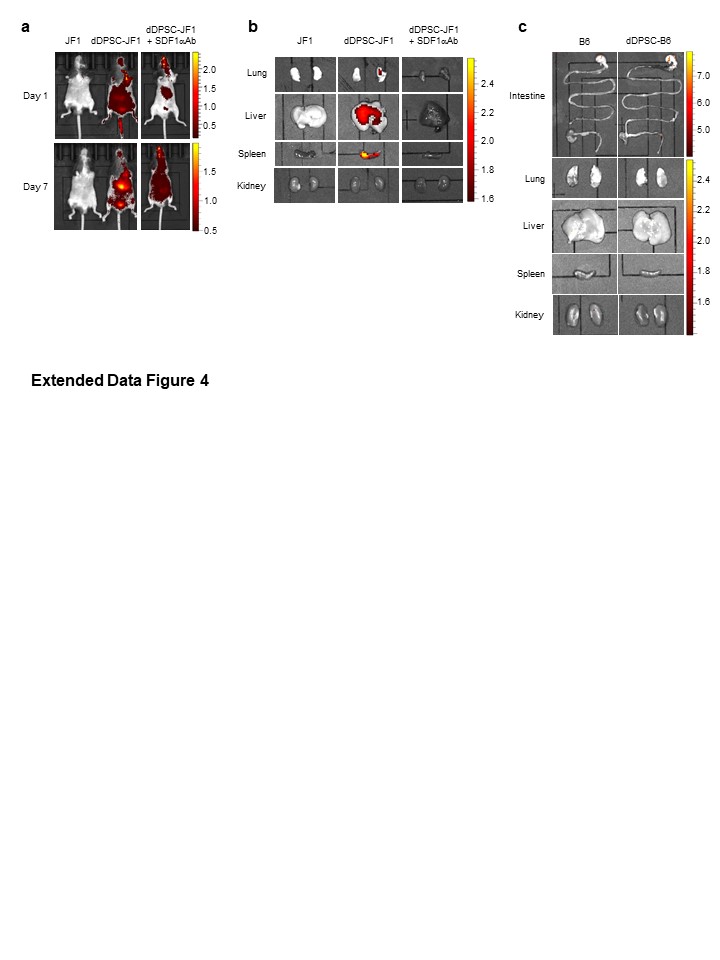
dDPSCs, BMMSCs or FBs (104 cells/g bodyweight in 100 µL PBS) were intravenously injected into JF1 mice at P11W. Age-matched B6 and JF1 mice intravenously injected with PBS (100 µL) were used as controls. Some JF1 mice were injected intraperitoneally with anti-SDF1α antibody (100 µg in 100 µL PBS; R&D Systems, Minneapolis, MN, USA) or PBS (100 µL) 24 hours before transplantation.
Mice were divided into five experimental groups: B6 (n = 10), (PBS-infused) JF1 (n = 10), dDPSC-transplanted JF1 (n = 10), BMMSC-transplanted JF1 (n = 10) and FB-transplanted JF1 (n = 10). Mouse bodyweight, water intake, food intake and stool number were measured weekly from P11W to P35W. Some mice were selected randomly for harvesting of organ tissues and peripheral blood at P23W (12 weeks after transplantation), and the intestine was dissected into lengths of ~15 mm.
Monitoring of transplanted cells
To reduce the background autofluorescence of standard chow, mice were fed with alfalfa-free Ivid#2 feed (Oriental Yeast) for 10 days before in vivo and ex vivo imaging. dDPSCs were incubated with DiR (Perkin Elmer, Waltham, MA, USA) for 30 min at 37°C and washed twice with PBS. Fluorescent dDPSCs (105 cells/g bodyweight in 100 µL PBS) were intravenously transplanted into recipient mice (P23W). PBS-infused mice were used as controls. Whole-body images and ex vivo images of organs were acquired 1, 7 and 20 days after transplantation using an IVIS Lumina III system (Perkin Elmer). In the experiment involving the injection of anti-SDF1α antibody into a JF1 mouse, ex vivo imaging was performed 18 days after dDPSC transplantation due to early death of the mouse. Colour images showing DiR fluorescence above a threshold level were superimposed on greyscale images of the mouse body or resected organ.
Peristalsis
Mice were fed with standard chow, which exhibits autofluorescence. The entire gastrointestinal tract was resected (P23W) after a 24-hour fast. The autofluorescence of the intestinal contents was measured from ex vivo images acquired with an IVIS Lumina III system (Perkin Elmer). A reduction in autofluorescence level was interpreted as an increase in peristaltic movements.
Nutritional status
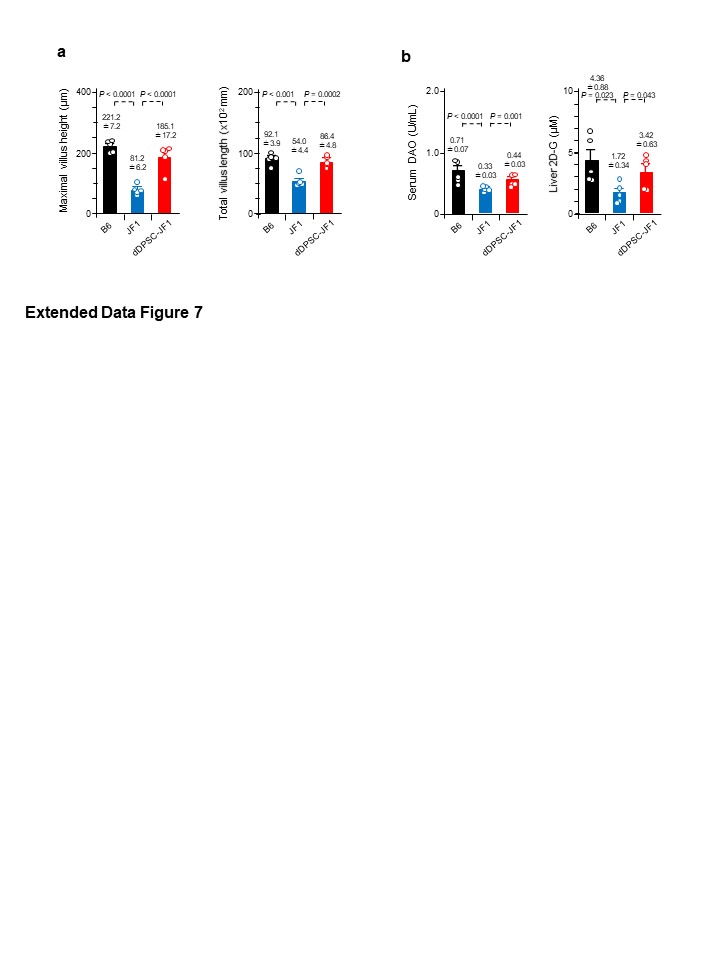
Improvements in nutritional status (hepatic glucose uptake and body weight) after dDPSC transplantation were used as surrogate markers of mucosal repair in the small intestine. Tibial bones were harvested from each experimental group at P23W, and the distal tibial epiphyses were imaged using a Skyscan 1076 micro-CT system (Skyscan, Kontich, Belgium). Bone mineral density (BMD) and bone parameters were estimated using CT-Analyzer software (Skyscan). BMD values were calibrated using hydroxyapatite phantoms with BMD values of 0.25 and 0.75 g/cm3 (Skyscan).
In vivo hepatic glucose uptake was measured in mice (P23W) given free access to water containing 1% 2-deoxy-D-glucose (2DG; Merck)45. Liver tissue samples were collected after 7 days. Hepatic 2DG content (an index of glucose storage) was assayed using a commercial kit (Cosmo Bio, Otaru, Japan) and plate reader (Multiscan GO; Thermo Fisher Scientific).
Histology and immunohistochemistry
Samples were fixed with 4% paraformaldehyde in PBS (pH 7.2) at 4ºC overnight, dehydrated, cleaned and embedded in paraffin. For histology, sections were stained with haematoxylin-eosin. For immunohistochemistry, sections were incubated first with 3% H2O2 in ethanol for 30 min to inhibit endogenous peroxidase and then with 10% bovine serum albumin in PBS for 60 min. Sections were treated with 0.01 M citrate buffer (pH 6.0 or 9.0) in a microwave processor (MI-77, Azumaya, Tokyo, Japan) for antigen retrieval. Sections were incubated with primary antibodies (Supplementary Table 2) at 4ºC overnight. Visualisation was achieved using an EnVision+ System (Agilent, Santa Clara, CA, USA). Sections were lightly counterstained with haematoxylin. For double-immunofluorescence staining, sections were incubated with primary antibodies (Supplementary Table 2) and then treated with the Opal 3-plex kit (Perkin Elmer). Sections were mounted onto slides using Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). Negative controls utilized isotype-matched antibodies instead of primary antibodies. Images were obtained using a BZ-X700 fluorescence microscope (Keyence, Tokyo, Japan). The number and area of ganglion cells in myenteric ganglia were estimated using Hybrid Cell Count software (Keyence)46–48.
Quantitative assays
Blood samples were incubated overnight at 4ºC and centrifuged at 1,500 rpm for 10 min at 4ºC to obtain serum. Tissue samples (including liver, lung and kidney) were homogenized in T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific) containing a proteinase inhibitor cocktail (Nacalai Tesque) using a Vibra-Cell ultrasonic processor (Sonic & Materials, Newtown, CT, USA). Total protein was measured using a protein assay (Bio-Rad Laboratories, Hercules, CA, USA) and normalized to the tissue sample weight.
Commercial kits were used to measure serum levels of creatinine (Creatinine Colorimetric Assay kit, Cayman Chemical Company, Ann Arbor, MI, USA), alanine aminotransferase and aspartate aminotransferase (Transaminase CII-Test kit, Wako Pure Chemicals). Commercial enzyme-linked immunosorbent assay (ELISA) kits were used to determine albumin, GDNF, IL-2, MIP-2, NGF, SCF and SDF1α protein levels in mouse tissues or serum (see Supplementary Table 3). The colourimetric assay and ELISA results were analysed using a Multiscan GO plate reader (Thermo Fisher Scientific).
Serum levels of IL-1α, IL-1β, IL-2, IL-5, IL-6, IL-12, IL-18, granulocyte-colony stimulating factor, tumour necrosis factor-α, interferon-γ and macrophage inflammatory protein-1β (MIP1β) were quantified using a Bio-Plex MAGPIX system and a Bio-Plex Pro multiplex cytokine assay system (Bio-Rad Laboratories).
RT-qPCR
Samples were treated with TRIzol (Thermo Fisher Scientific). RNA extracts were digested with DNase I (Promega, Fitchburg, WI, USA) and purified using the RNeasy Mini Kit (Qiagen, Venlo, Netherlands). Total RNA samples were treated with a Revertra Ace qPCR kit (Toyobo, Osaka, Japan) to obtain cDNA. cDNA samples were mixed with EagleTaq Universal Master Mix (Roche, Basel, Switzerland) and target-specific TaqMan probes (Thermo Fisher Scientific; Supplementary Tables 4 and 5). RT-qPCR was performed using a Light Cycler 96 system (Roche) under the following conditions: 50°C for 120 sec; 95°C for 600 sec; 45 cycles of 95°C for 15 sec and 60°C for 60 sec. Glyceraldehyde 3-phosphate dehydrogenase mRNA (mouse samples) and 18S ribosomal RNA (human samples) were used for data normalization.
Mouse intestinal microbiota
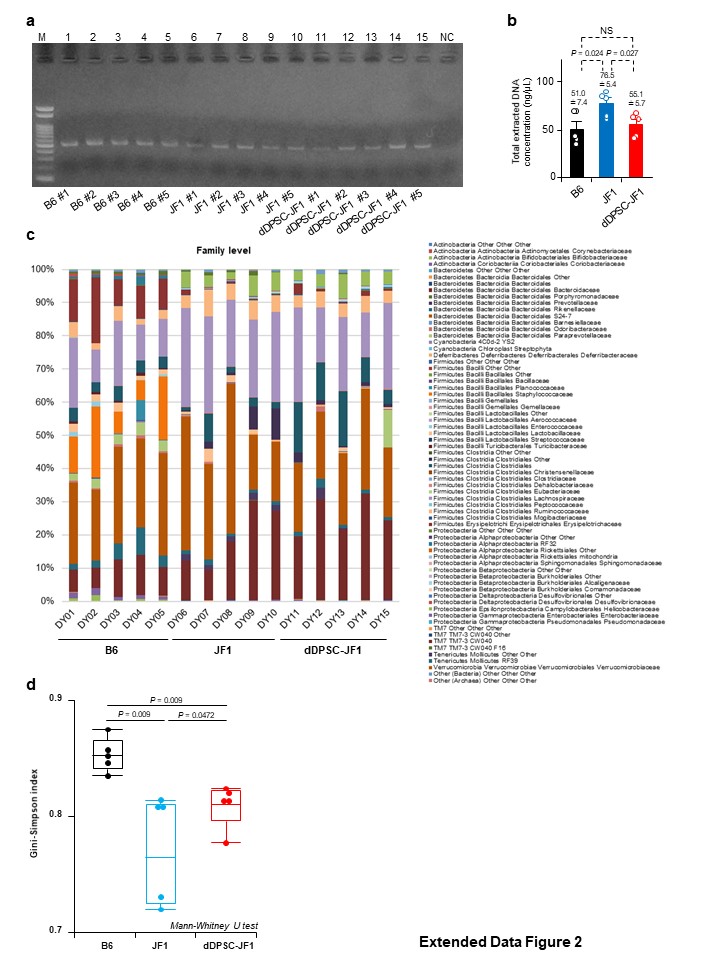
Stools were collected daily during P24W–P26W. Faecal bacterial DNA content was analysed by the Primary Cell Division of Cosmo Bio (Sapporo, Japan; protocol #151228A1 and #160125A1). Briefly, microbial DNA was extracted from the faecal sample using the Isospin Fecal DNA kit (Nippon Gene, Tokyo, Japan). The V3V4 region of the 16S rRNA gene was amplified by PCR (forward: CCTACGGGNGGCWGCAG; reverse: GACTACHVGGGTATCTAATCC) and sequenced using a MiSeq Deep sequencer and MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA). The sequence data were pre-processed and analysed using Flora Genesis software (Repertoire Genesis, Ibaraki, Japan). The R1 and R2 read pairs were joined, and chimera sequences were removed. Open-reference operational taxonomic unit (OTU) picking was performed using the 97% ID prefiltered Greengenes database and Uclust. Representative sequences of each OTU were chosen, and taxonomy assignment was performed using the Ribosomal Database Project (RDP) classifier and a threshold score ≥0.5. OTUs were grouped if their annotation was the same regardless of RDP score. Bacterial floral communities were characterised using the Gini-Simpson index (1/D)49,50.
Colonic mechanical activity
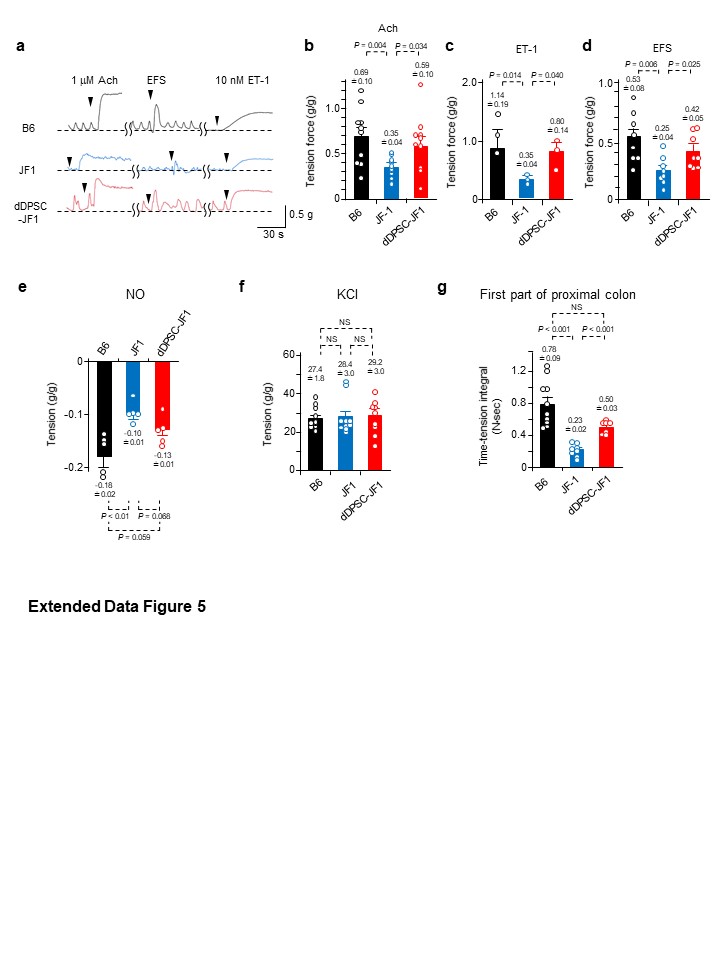
Mechanical responses to pharmacological agents and EFS were measured in 15-mm-long muscle strips isolated from the proximal colon. Macroscopically, the mouse proximal colon has two bulging regions; here we refer to the oral one as the first part and the anal one as the second part. The second part of the proximal colon was used in all experiments except those shown in Extended Data Figure 5g. The modified Krebs solution consisted of 121.6 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 1.2 mM Na2KH2PO4, 15.4 mM NaHCO3 and 11.5 mM D-glucose gassed with 5% CO2 and 95% O2 (pH 7.4 at 37ºC). The strips were mounted vertically in an organ bath maintained at 37ºC. Changes in isometric tension generated by the circular muscle were recorded using a force transducer (TB-612T; Nihon Kohden, Tokyo, Japan) and amplifier (AS1202; NEC, Tokyo, Japan) and analysed using LabChart and Scope software (ADInstruments, Bella Vista, NSW, Australia). Enteric nerve stimulation was achieved using EFS (50 V, 0.5-msec step pulses repeated at 5 Hz for 5 sec) applied by an electrical stimulator (SEN-3301; Nihon Kohden). The following pharmacological agents were used: 1 µM Ach (Wako Pure Chemicals), 10 nM ET-1 (Wako Pure Chemicals), 10 µM NO (Wako Pure Chemicals), 60 mM KCl, 1 µM atropine sulphate (Wako Pure Chemicals; to block the response to Ach) and 1 µM tetrodotoxin (Wako Pure Chemicals; to block the response to EFS). Tension development in response to EFS and pharmacological agents was normalized to the tissue weight and response to 60 mM KCl.
Colonic electrical activity
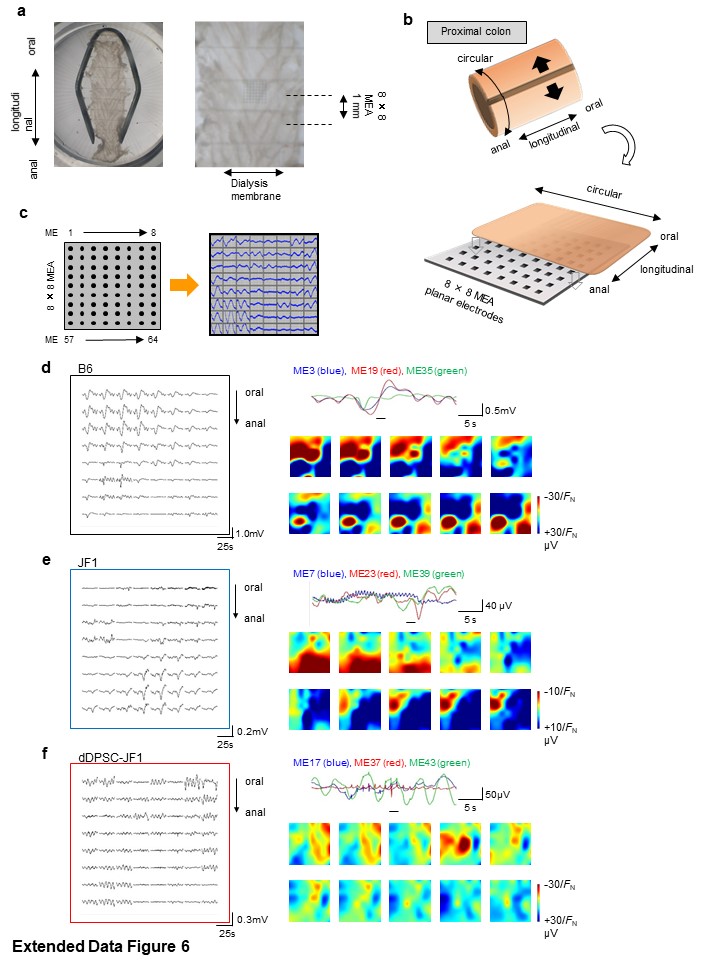
The spatiotemporal characteristics of proximal colon electrical activity were assessed using an 8×8 MEA recording system (Alpha MED Scientific, Ibaraki, Japan)51,52. Colonic smooth muscle samples were prepared by gently removing the mucosa. Each sample was mounted on an 8×8 MEA (interpolar distance, 150 μm) with the longitudinal muscle layer facing downward using a piece of dialysis membrane fixed with a slice anchor (SDH series, Harvard Apparatus Japan, Tokyo, Japan; Extended Data Figure 6). The 2-mL recording chamber was perfused (1–2 mL/min at 34°C) with modified Krebs solution containing HEPES/Tris pH-buffer instead of bicarbonate to prevent the development of microbubbles on the MEA. The sample was perfused with solution for ~30 min before recording was started, and perfusion was discontinued during recording to minimize electrical noise. A set of 8×8 field potentials were simultaneously recorded using a computer-controlled, multi-channel AC amplifier (low-pass filtering at 10 kHz) and 14-bit A/D converters (sampling rate of 20 kHz). The dynamic range of A/D conversion was usually ±1 mV. High-pass (0.1 Hz) filtering was applied to stabilize the baseline drift of the microelectrode potential.
A low-impedance MEA was employed to measure a wide range of electrical activity, including pacemaker potentials occurring at ~4-sec intervals and myoelectric complex-like potentials with rapid and slow components51. Each recording electrode was a square (~50 μm × 50 μm) made from platinum black nanoparticles, which increased the surface area by ~200-fold to 0.5 mm2. The capacitance (CME) and resistance (RME) of each microelectrode was 0.052 μF and 15 kΩ, respectively51,53, hence the impedance of the recording electrode at 0.1 Hz was estimated to be small enough [~31 MΩ = √ {1/(2π × 0.1 Hz × 0.052 μF)2 + (15 kΩ)2}] to follow oscillating potentials when compared to the input impedance of the multi-channel amplifier (100 MΩ at 0.1 Hz). The efficacy of electrical signal transmission (Tr) was estimated to be ~95% at 0.1 Hz [100 MΩ / √ {(100 MΩ)2 + (31 MΩ)2}]. Moreover, the low resistance (15 kΩ) of the electrodes was advantageous for reducing thermal noise (NT), which was calculated to be ~1.6 μV at the low-pass frequency (10 kHz) of the AC amplifier.
To generate pseudocolour images, the field potential data were thinned 100–1000-fold in the time domain and bandpass filtered at 0.25–30 Hz (Kaiseki Excel add-in software, Kyowa, Tokyo, Japan). At certain recording times, the 8×8 field potentials were interpolated by a spline function with 50 points between each potential using MATLAB (MathWorks, Natick, MA, USA)53. To reconstitute smooth potential images, the amplitude of the field potential in each ME was compensated by the linear spectrum at 0.1–15 Hz51. The upper and lower images corresponded to the oral and anal ends of the MEA recording region, respectively. Field potential videos were produced at a frame rate of 200 Hz.
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM) of at least three determinations. Inter-group comparisons were made using two-tailed Student’s t-tests for distinct samples (two groups), Mann-Whitney U-test or one-way repeated-measures analysis of variance (ANOVA) followed by the Tukey post-hoc test (three or more groups). Kaplan-Meier analyses and log-rank tests were used for survival analyses. P < 0.05 was considered significant. Analyses were performed using JMP 11 (SAS Institute, Cary, NC, USA).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}